Search times may vary depending on the computer hardware the Spectrum Mill workbench is running on, the size
of the database being searched, the restrictiveness of the search parameters, and the number of searches being
simultaneously performed. When two or more searches are being performed simultaneously, the searches slow noticeably.
In general, faster searches result from more discriminating search parameters: single species, narrow intact protein
MW range, 0 missed cleavages.
For MS/MS Search, see the section on Batch Size.
When you initiate a search, the search will still run to completion even if you click Stop on your browser. This is because the Stop process link terminates the main computation process, but not related processes. (Most Spectrum Mill programs run as perl scripts, which in turn run cgi programs.)
The unfortunate end result is that if you click Stop, change a parameter and resubmit the search request, each additional search becomes progressively slower because the server is running multiple searches.
However, there are several ways for you to terminate a search and resubmit without slowing the server:
Step 1. Stop the main process.
Step 2. Stop the related processes.
One Spectrum Mill search program can serve as a pre-filter for another search program. To accomplish this the hits (index numbers for matching database entries) from the first program are saved to a user-specified file. This file is then retrieved by the second program, and only those matching database entries are searched by the second program.
To save hits from MS Edman, mark the check box on the MS Edman form to Save hits to file and supply a file name. To create a subset database from these hits, go to the Protein Databases form and use the option to Create subset with indices from saved hits. Once you have created a subset database, you can search these hits with MS Edman or the other search programs. The spectra you search can reside in any data directory.
To save hits from MS/MS Search, use the Tool Belt page to Create saved results file. This file contains the saved accession numbers from validated hits (and so is like a mini-database of validated proteins). You can then use MS/MS Search to search spectra in the same data directory against these hits. For example, save hits from an identity mode search and then search the saved hits in homology mode.
When you mark the check box to Search previous hits in MS/MS Search, if you have not created a saved results file, then the search page automatically creates one for you. (On the form, be sure to select the database that you searched previously.)
Note that the check boxes for Disable quality filtering and Disable match filtering are available only if configured in SMglobals.js. See the server administration help for details.
When in Data Extractor you mark the check box to Disable quality filtering or in MS/MS Search you mark the check box to Disable match filtering, the software disables various filtering parameters within the Spectrum Mill workbench. At the Data Extractor level, the software disables the sequence tag length, spectral merging, and minimum signal-to-noise parameters. In addition, it no longer attempts to assign +2 and +3 charge states. At the MS/MS Search level, the software disables the minimum % SPI filtering, sequence tag length, and minimum signal-to-noise parameters. As a result, you observe greater sequence coverage, but the quality of the spectra that produced the additional coverage is poorer, making the results questionable.
CAUTION: Since the "Disable quality filtering" and "Disable match filtering" modes process a greater number of lower-quality spectra, the likelihood of false positives increases. We recommend you use this feature only when you work with a known sample where you desire increased sequence coverage (for example, a single-protein digest).
When you mark the check box to Disable quality filtering (sequence tag length = -1, no merging, attempt to assign charge +1 only), the following occurs:
* Note that Sequence tag length and Merge scans with same precursor m/z are reset internal to the software. The current settings on the form do not change, and they once again take effect when you clear the check box labeled Disable quality filtering.
When you mark the check box to Disable match filtering (SPI, STL, S/N filter), the following occurs:
** Note that Sequence tag length and Minimum scored peak intensity are reset internal to the software. The current settings on the form do not change, and they once again take effect when you clear the check box labeled Disable match filtering.
Spectrum Mill programs search sequence databases that are located locally on the server running the programs. The actual files searched are FASTA-formatted copies of the source database which contain minimal annotation. Search output typically contains a web-link into a fully-annotated version of the source database for each entry matched.
Spectrum Mill programs allow searching of the publicly-available genome and proteome databases listed below. However, nearly any sequence database in a suitable FASTA format can be set up for use by contacting the administrator of a Spectrum Mill server.
Note that the URLs for these databases may change over time, so you may need to search for the current URL. You may also check the Agilent Software Status Bulletin to see if there is an update for this file. To view this bulletin, click here.
Protein Databases
DNA Databases
Reasons to search particular databases:
Reasons NOT to search particular databases:
The local copy of the database being searched with the programs is subject to updating by the administrator of a Spectrum Mill server.
If you don't know the Latin taxonomic name for the species you're interested in, try: NCBI Taxonomy Browser
Species-limited searches in Spectrum Mill programs are performed by means of preliminary filtering of a database according to the user-designated species or collection of species. This species pre-filter is bypassed when the species is designated as All.
This species pre-filtering is imperfect because of the poor usage of taxonomy (standard species naming conventions) in the databases, AND the poorly standardized location of this information in the FASTA database formats used by Spectrum Mill programs.
Users who desire additional/changed species filtering capability should direct their local Spectrum Mill server administrator to the instructions To Add/Change Species Filter.
Species pre-filtering is implemented in Spectrum Mill programs by correlating the user-selected species name in the HTML form with the variety of pseudonyms for a particular species in the databases through behind-the-scenes access to a species alias list for each database used.
Below is a list of the variety of pseudonyms for Mouse.
| NCBInr | Genpept | Owl | SwissProt | |
|---|---|---|---|---|
MOUSE MUS MUSCULUS |
M. MUSCULUS M.MUSCULUS MOUSE MUS DOMESTICUS MUS MUSCULUS |
MUS MUSCULUS |
MOUSE MUS MUSCULUS MUS MUSCULUS (MOUSE) |
MOUSE |
Server Administrators can edit these alias lists without requiring access to Spectrum Mill source code. Note that this mechanism of pseudonym correlation allows for significant flexibility. For example, an alias can be created that includes a collection of species i.e. mammals, eukaryotes, prokaryotes, etc.
This is a list of some of the species definitions that are supplied with the Spectrum Mill workbench. Note that some definitions do not encompass all possible members.
| Species as listed in search program | Species included * | Common Name** |
|---|---|---|
| [FISH REPTILES] | CAENORHABDITIS ELEGANS | 'the worm' |
| DANIO RERIO | zebra fish | |
| XENOPUS LAEVIS | clawed frog | |
| [HUMAN MOUSE] | HOMO SAPIENS | human |
| MUS MUSCULUS | house mouse | |
| [HUMAN RODENT] | HOMO SAPIENS | human |
| MUS MUSCULUS | house mouse | |
| RATTUS NORVEGICUS | Norway rat | |
| [INSECT] | BACULOVIRUS | |
| DROSOPHILA MELANOGASTER | fruit fly | |
| ROACH LOCUST BEETLE | comprised of a number of species - see species.txt | |
| [MAMMALS] | BOS TAURUS | cow |
| CANIS FAMILIARIS | dog | |
| CAPRA HIRCUS | goat | |
| EQUUS CABALLUS | horse | |
| FELIS CATUS | cat | |
| GORILLA GORILLA | gorilla | |
| HOMO SAPIENS | human | |
| MACACA | macaques | |
| MUS MUSCULUS | house mouse | |
| ORYCTOLAGUS CUNICULUS | rabbit | |
| OVIS ARIES | sheep | |
| PAN TROGLODYTES | chimpanzee | |
| RATTUS NORVEGICUS | Norway rat | |
| SUS SCROFA | pig | |
| [MAMMALS MINUS HMR] | [MAMMALS] listed above, minus homo sapiens, mus musculus, and rattus norvegicus | |
| [NOT MAMMALS] | All species except those listed for [MAMMALS] | |
| [NOT PLANTS] | All species except those listed for [PLANTS] | |
| [NOT VIRUSES] | All species except those listed for [VIRUSES] | |
| PLANTS | ARABIDOPSIS THALIANA | thale-cress, mouse-ear cress |
| GLYCINE MAX | soybeans | |
| HORDEUM VULGARE | barley | |
| LYCOPERSICON ESCULENTUM | tomato | |
| MAIZE | Indian corn, corn | |
| NICOTIANA TABACUM | common tobacco | |
| ORYSA | rice | |
| ORYZA SATIVA | rice | |
| PISUM SATIVUM | pea | |
| SOLANUM TUBEROSUM | potato | |
| TRITICUM AESTIVUM | bread wheat | |
| ZEA MAYS | maize | |
| RODENT | MUS MUSCULUS | house mouse |
| RATTUS NORVEGICUS | Norway rat | |
| VIRUSES | HUMAN IMMUNODEFICIENCY VIRUS TYPE 1 | |
| HUMAN IMMUNODEFICIENCY VIRUS TYPE 2 | ||
| INFLUENZA A VIRUS | ||
| INFLUENZA VIRUS TYPE A | ||
| MEASLES VIRUS | ||
| PARAMECIUM BURSARIA CHLORELLA VIRUS | ||
| SIMIAN IMMUNODEFICIENCY VIRUS | ||
| TT VIRUS | ||
| VACCINIA VIRUS | ||
| ROACH LOCUST BEETLE | LEPDE | Colorado potato beetle |
| LOCMI | migratory locust | |
| PERAM | American cockroach | |
| SCHAM | American grasshopper | |
| SCHGR | desert locust | |
| SCHNI | vagrant locust | |
| LEPTINOTARSA DECEMLINEATA | Colorado potato beetle | |
| LEPTINOTARSA DECEMLINEATA=COLORADO POTATO BEETLES, SAY, PEPTIDE PARTIAL, 20 AA | ||
| LOCUSTA MIGRATORIA | migratory locust | |
| PERIPLANETA AMERICANA | American cockroach | |
| PERIPLANETA AMERICANA=AMERICAN COCKROACHES, PEPTIDE PARTIAL, 28 AA | ||
| SCHISTOCERCA AMERICANA | American grasshopper | |
| SCHISTOCERCA GREGARIA | desert locust | |
| SCHISTOCERCA NITENS | vagrant locust |
* These may have additional species aliases listed in species.txt.
* *Disclaimer: Much of this information was derived from the NCBI taxonomy database (
http://www.ncbi.nih.gov/Taxonomy/ ), which claims
to not be an authoritative source for nomenclature or classification. Please consult the relevant
scientific literature for the most reliable information.
Intact protein molecular weight (MW)-limited searches in Spectrum Mill programs are performed by preliminary filtering of a database according to the user-designated intact protein MW.
The intact protein MW pre-filtering is imperfect because sequences in protein databases often exist in pre, pro, and fragment forms.
The Protein Database program ALWAYS calculates the intact protein MW, according to the following constraints.
The Spectrum Mill workbench no longer supports filtering by protein pI in MS/MS Search.
Intact protein pI-limited searches in other Spectrum Mill programs are performed by preliminary filtering of a database according to the user-designated intact protein pI. This pre-filter is bypassed when the pI range checkbox All is marked.
The intact protein pI pre-filtering is imperfect because sequences in protein databases often exist in pre, pro, and fragment forms.
Spectrum Mill programs always calculate the intact protein pI, according to the following constraints.
Spectrum Mill server administrators can modify the pK values that are used to calculate the pI values. You should not modify the pKs for the standard amino acids unless a value has been determined to be wrong. You must remake the database index files using Protein Databases if you change the pK values.
Spectrum Mill server administrators may set the pK values for modifications when they define modifications (only necessary if the pK values are different from those of the unmodified amino acid). The Spectrum Mill workbench uses the pK values for the modifications to calculate the pI for peptides, but does not use them to calculate the protein pI values. The protein pI values are calculated when the database is indexed and they represent the pIs of the unmodified proteins.
Results from MS/MS Search can be filtered by required or disallowed amino acids. Filtering can be done at the Protein/Peptide Summary level.
The advantages of filtering at the Protein/Peptide Summary level are possible reduction of false positives and additional flexibility for results analysis. For example, you can generate one summary to review all results, then generate a second summary to more closely inspect a subset of results (e.g., phosphorylated peptides).
Protein/Peptide Summary provides a drop-down menus for amino acid filtering. Your server administrator can customize the options in the drop-down menus.
| Required AA | Description |
|---|---|
| any | No amino acid filtering |
| y | Show only peptides that contain phosphorylated tyrosine |
| s|t|y | Show only peptides that contain either phosphorylated serine, phosphorylated threonine, or phosphorylated tyrosine |
| s|t | Show only peptides that contain either phosphorylated serine or phosphorylated threonine |
| C | Show only peptides that contain cysteine, either unmodified or with a fixed modification |
| C|c | Show only peptides that contain cysteine, either unmodified, with a fixed modification, or with a variable modification |
| c | Show only peptides that contain cysteine with a variable modification |
| Disallowed AA | Description |
|---|---|
| none | No amino acid filtering |
| m|q | Show only peptides that do not contain oxidized methionine or pyroglutamic acid |
| m|q|s|t|y | Show only peptides that do not contain oxidized methionine, pyroglutamic acid, phosphorylated serine, phosphorylated threonine, or phosphorylated tyrosine |
| y | Show only peptides that do not contain phosphorylated tyrosine |
| s|t | Show only peptides that do not contain phosphorylated serine or phosphorylated threonine |
| s|t|y | Show only peptides that do not contain phosphorylated serine, phosphorylated threonine, or phosphorylated tyrosine |
| C | Show only peptides that contain cysteine, either unmodified or with a fixed modification |
| C|c | Show only peptides that contain cysteine, either unmodified, with a fixed modification, or with a variable modification |
| c | Show only peptides that contain cysteine with a variable modification |
Note! As of B.06.00, searching of DNA FASTA databases such as dbEST or custom databases (DN or DA prefix) are no longer supported. The DNA sequences must be converted to protein sequences. The FASTA protein header lines must correspond to one of the supported formats. See Updating databases
The termini of the matched peptides can be set to be consistent with the cleavage specificity of the enzyme used to generate the peptide. When you select No enzyme (not available in PMF Search or MS Digest), the matched peptides have no constraint on their termini. When you increase the maximum number of missed cleavages, you enable matching to sequences with uncleaved sites internal to the peptide.
For Spectrum Mill version A.03.02, the default number of missed cleavages was increased from 1 to 2 for MS/MS Search. In general, this leads to more identifications. However, for the unusual case of a homology search or a variable modifications search with many modifications against a full database, you may achieve more valid identifications with 1 missed cleavage than with 2 missed cleavages.
The option for the non-existent enzyme Trypsin/Chymotrypsin was created as a means to allow chymotryptic cleavages in trypsin digests. When you select this choice, it is important to increase the allowed missed cleavages. Increasing to 9 will result in only a marginal increase in search time.
It is possible to combine the rules for two or more enzymes by adding options to the Enzyme item on
the HTML form. For example adding the option:
<OPTION> CNBr/Trypsin/Asp-N
would combine the cleavage rules for CNBr, Trypsin and Asp-N.
It is possible to mix N-terminal cleavage rules with C-terminal ones in this way.
Spectrum Mill server administrators can edit the existing enzyme cleavage rules or add new ones.
The links in program output are intended to easily facilitate user access to obvious sources of additional information about proteins or peptides matched or under study. Some of the default parameters of these links can be changed by Spectrum Mill server administrators.
Server administrators can change the default parameters in the HTML links from the:
accession number
MS Digest index number
peptide sequence
elemental composition
The database accession number in the search results has a HTML link to retrieve the complete entry, including comments from a remote database. In order for this link to be created, the programs need to know the URL for the remote database. Users who desire links to different fully annotated databases, or who find links to a particular database to be defective, should contact their local Spectrum Mill server administrator.
Server Administrators can change the default address of links from accession numbers in program output without requiring access to Spectrum Mill source code.
The MS Digest index number in the search results has an HTML link to retrieve a listing of all the masses and sequences of peptides that can be produced by digesting the matched protein with the designated enzyme. If No enzyme was designated in the search parameters, then Trypsin is supplied in this HTML link. The number of missed cleavages is set to 2 unless a higher number was designated in the search parameters.
Server administrators can change the HTML link from the MS Digest index number in the search results.
If the accession number link marked Coverage Map in the PMF Summary detailed results is clicked, then the protein display at the top of the MS Digest report has the matching peptides highlighted.
The peptide sequence in the search results has an HTML link to MS Product for retrieving a listing of the theoretical fragment-ions that may be formed in an MS/MS experiment. The default set of ion types supplied in this link corresponds to those expected to be formed in post-source decay (PSD) experiments.
Server administrators can customize the HTML link from the peptide sequence in the search results.
The elemental composition in the search results has an HTML link to MS Isotope for retrieving a listing and visualization of the isotopic distribution corresponding to the composition.
Server administrators can customize the HTML link from the elemental composition in the search results.
The Spectrum Mill workbench supports the following modification types:
When you select Fixed/Mix Modifications:
When you select Variable Modifications:
The following table lists descriptions of some of the modifications. For more details about the modifications that are currently displayed on your server, click the Details button within the Choose... dialog. For information about additional modifications, use the Tool Belt utility to list modifications details.
Your server administrator can add custom modifications.
| Modification | Comment |
|---|---|
| Acetyl (ProtN-term) | Acetylation of any N-terminal amino acid. If methionine is present at the N-terminus, the methionine is removed and the next amino acid (the new terminus) is acetylated. |
| Acrylamide | Acrylamide modification usually means that there was no deliberate attempt at alkylation. Cysteines in a protein/peptide become alkylated during their exposure to the acrylamide in the SDS gel. |
| Amide | Modification of the carboxy group on the C-terminal amino acid. Common in peptide hormones. |
| Carbamidomethylation | The alkylation of a cysteine residue upon its reaction with iodoacetamide |
| Carbamyl | Modification results from treating a sample with urea. Since modification may be incomplete, see also Carbamyl-mix (below). |
| Carbamyl-mix | Since this modification results from treating a sample with urea that can result in incomplete modification, MS/MS Search searches each spectrum twice, once as modified and once as unmodified. The results are merged so that a single output report is generated. |
| Carbamylated lysine | Results from exposure of sample to urea. |
| Carboxymethylation | The alkylation of a cysteine residue upon its reaction with iodoacetic acid |
| clCAT | This ICAT reagent specifically alkylates cysteine residues. In addition to its reactive thiol group it also consists of a biotin affinity tag used to isolate ICAT-labeled peptides and a linker for the incorporation of the 13C stable isotopes. |
| DTT | The alkylation of cysteine residues with DTT. This usually occurs during a reduction step when excess DTT (cyclic, reduced) reacts with cysteine-containing peptides/proteins. |
| Guanidination | Guanidination results from the application of O-methylisourea to lysines to create homoarginine. This is primarily used to improve the MS sensitivity of lysine-containing peptides in trypsin digests. It is also used to block the amine group of lysines for reagents such as propionyl that are intended to modify only the N-terminus. |
| ICAT | This ICAT reagent specifically alkylates cysteine residues. In addition to its reactive thiol group, it also consists of a biotin affinity tag used to isolate ICAT-labeled peptides and a linker for the incorporation of deuterated stable isotopes. |
| iTRAQ | Protein digests are treated with a set of four isobaric reagents, which label all primary amines, including all N-termini and lysine. The labeled peptides have identical masses in MS mode, but produce diagnostic low-mass MS/MS signature ions, which allows simultaneous quantitation of up to four different samples. |
| Lys imidazole | Lysine residues within peptides react with 2-methoxy-4,5-dihydro-1H-imidazole, converting them to their 4,5-dihydro-1H-imidazole-2-yl derivatives. The resulting peptides ionize more efficiently. |
| Methyl Ester | This modification results in conversion of all carboxylic acid moieties to methyl esters. Thus selecting this modification also triggers Spectrum Mill programs to automatically redefine the mass of Asp and Glu residues to their methyl ester forms. |
| N15 | Used in experiments where cells are grown with a source of 15N. |
| Neuropeps | No longer supported because change of enzyme during a search cycle is not supported. |
| O18 Free Acid (double modification) |
Results from the digestion of a peptide bond in the presence of 18O water. Upon cleavage, the water is incorporated into the newly formed carboxyl ends. |
| PTC (N-term only) | Phenylthiocarbamyl modification of N-terminus. Lysines are treated as unmodified (i.e., lysines were blocked). |
| Propionyl | Protein digests are treated with propionic anhydride and undergo an acylation reaction in which the peptides are N-terminally modified. When you select this modification, you usually select guanidination as well. This reagent comes in two forms (D0 and D5) so it can be used for quantitation. |
| Pyridylethylation | The alkylation of a cysteine residue upon it reaction with vinylpyridine. |
| Pyroglutamic acid | Naturally occurring or chemically induced modification. During proteolytic digestion, N-terminal glutamine residues can sometimes cyclize to form the pyroglutamyl derivative. |
| SILAC | SILAC = stable isotope labeling of cells in culture. Cells are grown in media that contain amino acids that are labeled with various combinations of 13C, 15N, and D. |
Some Spectrum Mill programs allow the use of a user specified amino acid for which you must supply the elemental composition. To specify the user defined amino acid in a peptide or protein sequence use the letter u (lower case). The default elemental composition for the user defined amino acid is that of glycine.
Spectrum Mill programs expect the mass input values to represent the actual m/z values measured on a
mass spectrometer. Thus protons - H
Monoisotopic: only the lowest common isotope for each element is used in the mass calculations:
Monoisotopic no e- correction: same as Monoisotopic, except that for H+, the mass calculation uses the mass of a hydrogen atom rather than the mass of a proton. Use this Mass Type for MALDI QSTAR instruments when the mass calibration is done in such a way that there is no correction for the loss of the electron for protonated species.
Note: Prior to Spectrum Mill workbench version A.03.02, the Monoisotopic mode was really Monoisotopic no e- correction mode.
Average: All isotopes for each element are used, with their abundances reflecting their "normal" proportion in the biosphere.
This option is used to limit the maximum number of hits displayed. For example, if the maximum number of reported hits is set to 50 and there are 100 hits, then only the first 50 hits are displayed.
This option allows a user-defined comment or sample identifier to be added the output.
Searches can be restricted to matching sequences containing particular amino acid(s) by checking the appropriate boxes. This information can be derived from the masses of immonium and related low-mass ions or high-mass ions indicating side-chain losses from the precursor ion. The programs do not actually use the mass values but instead filter the matched sequence for the presence of the designated amino acid(s).
MS/MS searches handle immonium ions differently.
For the variable instrument_charges_certain:
Both 1 and 2 assign charge based on isotope spacing, with 2 being less aggressive. The choice of 1 or 2 depends on the expected ion statistics in your data and your tolerance for noise. With MS/MS Search, noise causes fewer problems because peak intensity contributes to the score. With PMF Search, noise can be more problematic because a peak contributes to the score regardless of intensity. Unassigned peaks reduce the score in both programs.
instrument_charges_certain = 2 (= if determined)
instrument_charges_certain = 1 (= yes)
instrument_charges_certain = 0 (= no)
Click the Select ... button to select a data directory.
In the Select Data Directory -- Web Page Dialog, click the data directory or directories you wish to select. Note that data directories are indicated by a rectangle within the folder icon.
| Icon | Used to designate | Behavior when you click |
|---|---|---|
| Folder | Directory that does not have data files directly beneath it | Directory not selected |
| Folder with rectangle | Directory that DOES have data files directly beneath it | Directory selected |
| Folder with line spectrum | Data file | Entire directory containing data file selected |
You can select multiple data directories in some forms, such as Data Extractor, MS/MS Search, Autovalidation, and Protein/Peptide Summary. When selection of multiple data directories is possible, use CTRL-click to select the second directory. (SHIFT-click does not work.)
Make Default
Mark this check box to have the software remember your data directory even after you close your web browser window. Note that the software keeps track of defaults from forms that allow multiple file selection separately from defaults for forms that allow single file selections. So if you make a directory or directories the default in Protein/Peptide Summary (a multi-directory form), these will not be retained as defaults in MS/MS Search (a single-directory form). However, the single-directory defaults (e.g., from MS/MS Search) will transfer to the multi-directory forms (e.g., Protein/Peptide Summary).
Any time you select a directory, the Spectrum Mill workbench automatically loads that directory in other forms used to process the same type of data. However, unless you mark this check box, it will not remember the data directory after you close your browser window. It will instead remember the last directory for which you marked this check box.
The Save Settings button saves all form settings, except for the data directory, between browser sessions.
If some or all of your data directories fail to appear on the list, see the tips.
The Load Parameters dialog box allows you to load a parameter file whose previous settings you saved.
The Save Parameters dialog box allows you to save a parameter file that you have created one of the Spectrum Mill pages.
Click the Choose... button to display the Choose Modifications dialog.
When you select modifications, some combinations are disallowed and are unavailable. The following table describes the available/unavailable combinations. The software permits only the appropriate combinations, so you do not need to consult the table to make the proper selections.
| For the same amino acid, can I choose: | |||
|---|---|---|---|
| Modification initially chosen | Another fixed mod? | A variable mod? | Another mixed mod? |
| Fixed, not metabolic | no | yes | no |
| Fixed, metabolic | yes | yes | no |
| Mix - for quantitation, not metabolic | no | no | no |
| Mix - for quantitation, metabolic | yes | yes | no |
| Mix - not for quantitation | no | yes | no |
For more information, see Modifications.
Reset
The Reset button resets settings for the Choose Modifications dialog to those last saved with the Save Settings button on any Spectrum Mill form that allows you to choose modifications. If the Save Settings button has not yet been clicked, the software resets the settings for the Choose Modifications dialog to the Spectrum Mill defaults.
Details
The Details button lists details about the modifications that are currently displayed on your server. To view information about both current modifications and additional modifications that could be configured on your server, see the information about the Tool Belt utility to list modifications details.
To import data into Excel or LIMS:
In Protein/Peptide Summary, Excel Export generates a specific .ssv (semicolon-separated variable) file for each summary mode:
| Summary Mode | Generated File |
|---|---|
| Peptide | peptideExport.N.ssv |
| Protein Summary | proteinSummaryExport.N.ssv |
| Protein - Protein Comparison Columns | proteinProteinCentricColumnsExport.N.ssv |
| Protein - Protein Comparison Redundant | proteinProteinCentricRedundantExport.N.ssv |
| Protein - Sample Centric Rows | proteinSampleCentricRowsExport.N.ssv |
N is incremented to guarantee a unique name for each file.
In PMF Summary, Excel Export generates a .ssv file of the form: msfitSummary.N.ssv, where N is incremented with each summary request.
The Spectrum Viewer is a tool to visualize the sequence information contained in MS/MS spectra and to evaluate spectral interpretations from MS/MS database searches or Sherenga de novo sequencing. To manipulate the Spectrum Viewer, see the descriptions below.
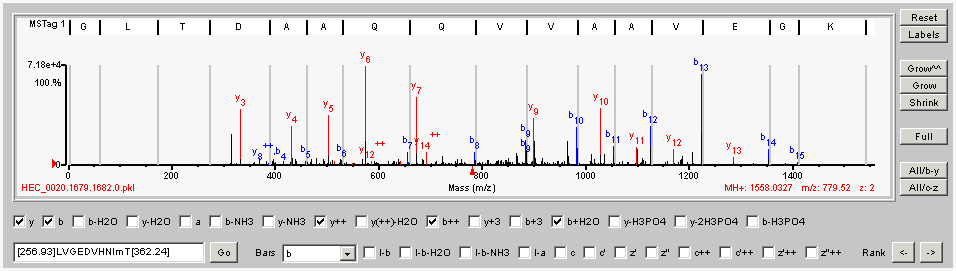
Hint: For Bars, choose an ion type to quickly see how well the sequence from the summary table matches the spectrum.
In the Spectrum Viewer, the software color-codes the ions as follows:
Use the buttons on the right side of the Spectrum Viewer to manipulate the display. The buttons do the following:
| Button | Function |
|---|---|
| Reset | Resets the spectrum to the original x- and y-axis values |
| Labels | Toggles among different peak labeling options. These are the default of interpreted peaks (b, y, etc.), interpreted peaks plus mass labels of all peaks, interpreted peaks plus mass labels of interpreted peaks, and no labels. To see the spectrum without the peak interpretations, click the red file name under the sequence at the top of the Spectrum Viewer. Now click the Labels button to turn the mass labels on and off. Note that you will need to re-display the spectrum to display the interpretations again. |
| Grow^^ | Expands the spectrum by ten times in the vertical axis |
| Grow | Expands the spectrum in the vertical axis |
| Shrink | Reduces the spectrum in the vertical axis |
| Full | Displays the spectrum with the full x-axis values. If you see a black arrow at the lower right-hand corner of the spectrum, this means that some of the spectrum is not displayed because there were no significant peaks in that region. Click the Full button to display the full x-axis range of the spectrum. |
| All/b-y | Toggles marking of check boxes for b- and y-ions in the first row under the spectrum and labels peaks accordingly. The toggle either marks all the check boxes or resets to a default subset. |
| All/c-z | Toggles marking of check boxes for c- and z- ions in the second row under the spectrum and labels peaks accordingly. The toggle either marks all the check boxes or resets to a default subset. In addition, the All/c-z button labels precursor ions that have reduced charges, as are typically observed in ETD spectra. When you analyze ETD data, you must click the All/c-z button to see the appropriate labels on the fragment ions. |
| Feature | Function |
|---|---|
| Sequence | Spectra are annotated with interpretations based on the sequence shown in white. When both MSTag and Sherenga sequence bars are displayed, click the sequence for which you wish to see annotations. |
| File name | To see the spectrum without the peak interpretations, click the red file name under the sequence at the top of the Spectrum Viewer. Now click the Labels button to turn the mass labels on and off. Note that you will need to re-display the spectrum to display the interpretations again. |
Mark check boxes for the ion types you want to see labeled in the spectrum. If you mark check boxes but do not see any labels, click the Labels button to turn the labels back on.
Use these features to manipulate the display. These features do the following:
| Feature | Function | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Go | To display a sequence above the spectrum, type the sequence in the box to the left and then click
the Go button. The sequence in the box is initially set to a default sequence. Mass gaps shown in brackets indicate portions of the spectrum where there was insufficient fragmentation to provide an amino acid sequence. You can enter mass gaps in the middle of the sequence as well as at the ends. Note that in addition to single-letter capitalized abbreviations for the 20 amino acids, you can type the following lower-case abbreviations for modified amino acids:
The variable modifications kmqsty are defined by default for the Spectrum Viewer. But if in MS/MS Search, you specified a different variable modification for K, M, Q, S, T, or Y (for example, guanidination of K), then that modification is used instead. That is, the default ‘kmqsty’ modifications are defined in addition to whatever variable modifications you selected, but any selected variable modifications have priority. You can use the Rank arrow buttons (<- and ->) to go from the sequences that were identified by MS/MS Search or Sherenga de novo Sequencing to the sequence that you typed. For MS/MS Search, the arrow buttons cycle between the peptide from the highest-scoring MS/MS search result and the sequence you typed. For Sherenga, the arrow buttons cycle through all the Sherenga result sequences. If you add a custom sequence, the software appends it to the list of Sherenga sequences that can be cycled. |
||||||||||||||
| Bars | Select an ion type to highlight. This allows you to more easily visualize how the ions align with the amino acid sequence that is displayed at the top of the spectrum viewer. | ||||||||||||||
| Check boxes labeled l-x | Mark these if you want to label peaks with amino acid sequences combined with common losses. | ||||||||||||||
| Check boxes for c- and z-ions | Mark check boxes for the ion types you want to see labeled in the spectrum. If you mark check boxes but do not see any labels, click the Labels button to turn the labels back on. | ||||||||||||||
| Rank | Click arrows to display additional Sherenga results. Note that when both MSTag 1 and Sherenga 1 sequence bars are displayed in the Spectrum Viewer, you must first click the Sherenga sequence bar and make sure it turns from gray to white before you click the Rank arrows. |
Use the cursor to expand a portion of the spectrum in the x-axis. Move your mouse over the spectrum. When a crosshair is displayed, select the portion of the spectrum you wish to expand.
Double-click the spectrum or click the Reset button to return to the original display.
The red triangle to the left of the y-axis indicates the threshold for peak labeling. Click anywhere on the y-axis to change this threshold.