Workflow Automation in Spectrum Mill
Table of Contents
Introduction to Workflow Automation
Spectrum Mill B.04.00 introduced workflow automation via a Service Request Manager(SRM), which allows you to place a series of tasks into a queue;
then the SRM executes them in the proper order. You can automate the tasks involved in a typical data
analysis for MS/MS data files from protein digests:
- Data Extractor
- MS/MS Search
- Autovalidation
- Quality Metrics
- Protein/Peptide Summary
The SRM Workflow automation was designed and developed with the following goals in mind:
- Provide automated workflow, linking multiple tasks (Extraction, MS/MS Search, Autovalidation, Protein/Peptide Summary Report).
- Handle dependencies of later tasks on previous tasks.
- Server load balancing, queue launches next workflow/task when a free processor is available.
- Asynchronous operation of SRM with no need for user to keep a web browser window open.
- Enable viewing of Queue Status and Completion log.
- Simple to use for experienced SM users, with minimal changes to existing user interface (UI) html forms.
- Same UI for both parameter review of individual tasks in an automated workflow and manual operation of individual tasks.
- Task queueing handles both automated workflows of multiple tasks and manually launched individual tasks.
Overview of Spectrum Mill modules and workflow
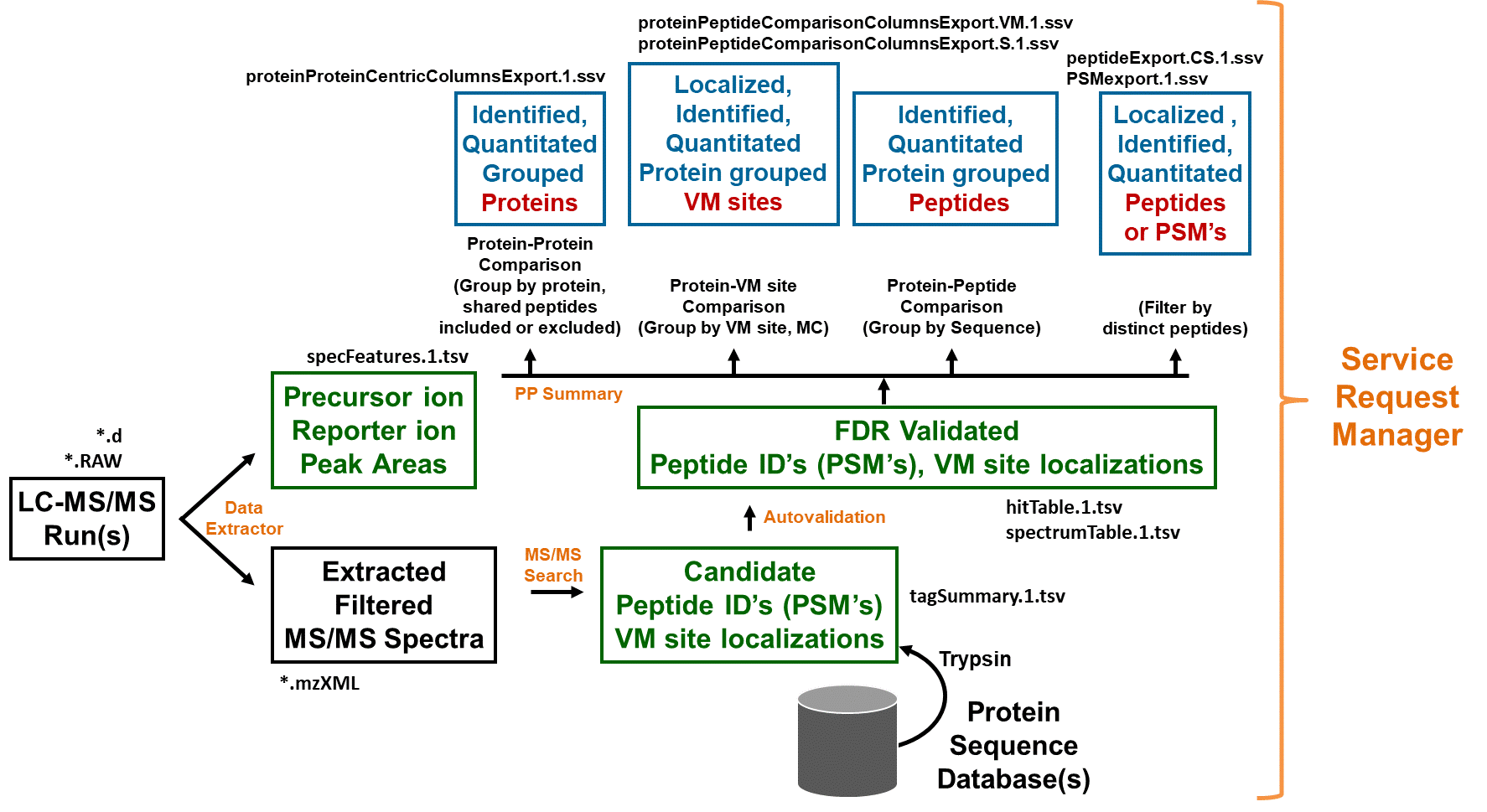
Parameter Files
The first step to set up an automated workflow is to save and name parameter sets you want to use for each task in your workflow.
Most Spectrum Mill forms allow you to save and load parameter files associated with the form. For example, Data Extractor, MS/MS
Search, Autovalidation, Quality Metrics, de novo and Protein/Peptide Summary all include buttons to save settings in parameter files, and
to load settings from parameter files. In nearly all cases, you use the parameter files to build and execute workflows.
Peptide Selector and MRM Selector are exceptions. For these programs, it is
convenient to save parameter files, but you can not use them in a workflow.
Parameter files are stored on the Spectrum Mill server under the folder \SpectrumMill\millauto. Only one level
of subfolder is allowed. We recommend that you assign subfolders to users or types of experiments or studies.
Each task type stores (in a cookie) the last parameter file that you selected for that task. The program shows
the selected parameter file in red in the title portion of the form:

Workflow Automation
Workflow automation user interface (UI) elements consist of a Workflows form, an Edit Workflow form, and a Request Queue / Completion
Log viewer. You access these forms through the Process Automation Tools section of the Spectrum Mill
home page. Workflow automation allows the Service Request Manager(SRM) to execute a workflow consisting of an ordered list of tasks on one or
more data folders using the same set of parameters for all data folders. Each task in the workflow must be accompanied by a parameter file which
specifies the settings for the task. Consequently, all data folders processed together should contain the same
data types for each task (for example with Extraction, all .raw, all .d files, etc.). When the SRM executes a workflow,
it adds the tasks to a Request Queue, and executes them in order for a particular data folder. If you select
multiple data file folders before you start the workflow, the SRM will perfrom the Data Extraction and MS/MS Search tasks in each folder in parallel.
With a typical multi core CPU configuration, the SRM processes a task on each logical processor,
up to the available processors. Certain tasks (Xtraction, MS/MS Search and de novo Sequencing) are launched for a data directory but execute in a series of
batches consisting of subsets of LC-MS/MS runs (Xtraction) or subsets of MS/MS spectra (MS/MS Search and de novo Sequencing) within the directory. These batches are
processed in parallel as sub-tasks on available processors when the max CPU option is checked on the workflow form or the individual task's form.
After a search is complete, autovalidation is performed independently for each folder unless "Group
proteins across ..." is marked. After autovalidation, results for all folders are summarized together by P/P Summary.
Note: If you want to have an autovalidation task using protein grouping process multiple directories independently, then you must separately
check and execute each one in the Data Directories section.
The Workflows and Edit Workflow forms allow you to view an individual task’s form in read-only mode, with
the parameters shown for the parameter file you select.
Service Request Manager(SRM)
In order to process workflows Spectrum Mill uses a Service Request Manager (SRM), implemented as a Windows service, to maintain
and operate a Request Queue, as illustrated below.
Overview of the SRM functionality
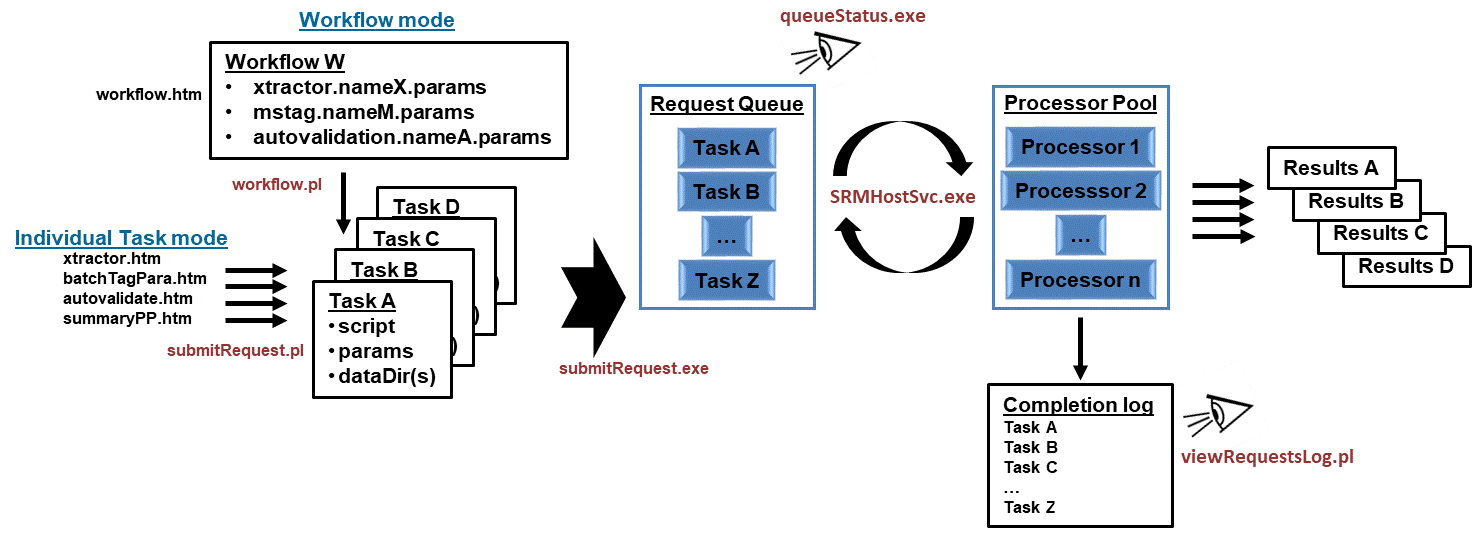
Together a Windows service, 2 programs, 4 scripts, the SRM configuration file, a log file, and UI generated parameter files for
workflows/tasks listed in the table below constitute the infrastructure of the SRM portion of Spectrum Mill.
| \\SpectrumMill/millsrm/ |
\\SpectrumMill/millscripts/ |
\\SpectrumMill/millauto/ |
- SRMHostSvc.exe
- submitRequest.exe
- queueStatus.exe
- SMSRM.Config
- requests.log
|
- workflow.pl
- submitRequest.pl
- queueStatus.pl
- viewRequestsLog.pl
|
- workflow.*.tsv
- xtractor.*.params
- mstag.*.params
- autovalidation.*.params
- ppsummary.*.params
- qualityMetrics.*.params
- sherenga.*.params
- specsummary.*.params
- specMatcher.*.params
|
Processor Utilization
The number of parallel tasks is limited by the number of (logical) processors on the server. By default, the Spectrum Mill installer
configures the SRM limit to be one less than the number of (logical) processors. For
example, a single CPU quad-core system with hyperthreading enabled should have 8 logical processors and be configured to allow the SRM to concurrently use 7.
You can edit a configuration
file to alter the maximum number of parallel processes. As
a general rule, you should have enough RAM to support each parallel process, at 2 Gb per process.
For more information, see Multicore (Maximize CPUs) Data Extraction.
See Stop and restart the SRM service in the event of reconfiguring the amount of system memory, or number of CPUs.
How/where to find the Results
Results for most Spectrum Mill modules will be placed in the same individual data directory as the original LC-MS/MS data files that
were selected for analysis. For tasks like Protein/Peptide Summary that combine results from multiple directories, the results will be placed
in the first directory selected in the input form.
SRM troublehooting aids
When a task is executed through the SRM, the output that was formerly output to a Results pane (typically the right-side
or below the form for the individual task) in Spectrum mill versions prior to 4.0, is now redirected to a task-generated HTML log file located in the data folder.
If the task (for example, Autovalidation or P/P Summary) uses multiple data directories, then the redirected output HTML is located in the first data directory in the list.
This is the same behavior as with the result tables produced for the Excel export option.
these redirected task-generated log files and are particulary useful when troubleshooting errant performance.
Redirected task-generated output logs produced during task execution The files have names like:
xtractorFinnigan.190101135721.1901P.htm
mstag.190101135723.1907P.htm
validate.190101135724.1909.htm
sherenga.190101135722.1903P.htm
qualityMetrics.190101135725.1910.htm
When tasks are launched from an individual task form for execution via the SRM, the links shown below
are present in the SRM acknowledgment output written to the lower pane. That output is redirected to an HTML file when
the tasks are executed as part of a workflow.
Redirected SRM acknowledgment of task submission to SRM request queue The files have names like:
xtractorFinnigan.190101135721.1902.htm
mstag.190101135723.1908.htm
sherenga.190101135722.1904.htm
To access the task-generated output log results via a web browser, look for these links in the SRM acknowledgment lower pane or redirected HTML output file:
- Link to Results - displays the output html (up to whatever content is available).
- Monitor Results - shows a window that continually updates as as a task proceeds.
(If you get too many Monitor Results windows running at once, you can use the Tool
Belt to stop the process.)
- View Request Queue - shows the Request Queue
See details about the Request Queue and Completion
Log, and how you can use them to view parameters and results output.
Workflow mode vs Individual Task mode
You can execute portions of a workflow one task at a time, by lanching requests directly from an individual task’s form.
In individual task mode, SRM Workflow automation remains dependency-aware and ensures that all tasks in a data folder
are processed in the proper order. As long as no dependencies exist, the tasks for different data folders execute in
parallel (up to the number of available CPUs) for extraction and search. When dependencies do exist (for example,
an MS/MS search cannot start until data extraction is complete), then the SRM ensures that the tasks execute in the
correct order and at the proper time.
In practice, certain combinations of tasks are well suited to workflow mode execution
(Data Extraction, MS/MS Search, and Autovalidation) and might most often be run that way. When all individual tasks are performed on a
a single directory the combination of them will tend to be Workflow friendly.
For some tasks it will tend to be convenient to simply run them in individual task mode. Examples include Autovalidation in
Protein polishing mode, and Protein/Peptide Summary in Protein Comparison mode when run across multiple data directories,
particularly for projects where the data in each directory is generated day or weeks apart.
When doing data analysis method development it will often be more convenient to run in individual task mode.
Bypassing the SRM
Before the SRM was developed all Spectrum Mill tasks could be executed by a user via direct connection between an
individual task's form and its script. This continues to be the case, and amounts to bypassing the SRM. For tasks
where users seek to generate tables of output in Excel Export format and primarily be viewed by with external tools,
bypassing the SRM would be unlikely to be beneficial.
However, for tasks such as Protein/Peptide Summary and Spectrum Summary that can produce HTML output with interactive
links, bypassing the SRM can be preferable. To enable this, check neither the Queue request checkbox nor the Excel Export checkbox
on the form. Example use cases include when one's primary goal is to:
- Inspect individual PSMs by following links from individual peptides to visualize MS/MS spectra with annotated fragment ion type assignments.
- Inspect the peptide coverage of the isoforms detected in protein groups/subgroups by following links from the protein group number to visualize the detected peptides highlighted in an alignment of the protein sequences.
Note: Although the forms for Data Extractor, MS/MS search, de novo Sequencing
no longer allow the SRM to be bypassed, the accompanying PERL scripts can still be run without the SRM. Instead of a Queue request checkbox,
the forms have a hidden variable that forces the feature to always be enabled when using the form.
Bypassing the User Interface with command line execution
KRC 1/4/2019 left off editing here
The main PERL scripts in Spectrum Mill are capable of receiving their parameters two different ways:
- Via Common Gateway Interface (CGI), with parameters intended to be passed from their web forms to the scripts via a web server.
Typically this is a Microsoft Windows operating system running Microsoft Internet Information Services (IIS).
- Via a Command Line Interface (CLI). This eliminates the need for a web server.
Command line syntax for launching Spectrum Mill PERL scripts is shown below:
each of the below is of the form:
>scriptName.pl paramsFilePath dataDirectoryPaths (space-delimited)
with detailed attention given to the pathes to the working directory, script, params and data directories.
E:\\SpectrumMill>millscripts\runXtractor.pl millauto\xtractor.name.params dataDir1 dataDir2 ... > msdataSM\dataDir1\xtractor.CLItest.htm
E:\\SpectrumMill>millscripts\batchTagPara.pl millauto\mstag.name.params dataDir1 dataDir2 ... > msdataSM\dataDir1\mstag.CLItest.htm
E:\\SpectrumMill>millscripts\validateTable.pl millauto\autovalidation.name.params dataDir1 dataDir2 ... > msdataSM\dataDir1\validate.CLItest.htm
E:\\SpectrumMill>millscripts\batchSherenga.pl millauto\sherenga.name.params dataDir1 dataDir2 ... > msdataSM\dataDir1\sherenga.CLItest.htm
E:\\SpectrumMill>millscripts\meterMaid.pl millauto\qualityMetrics.name.params dataDir1 dataDir2 ... > msdataSM\dataDir1\qualityMetrics.CLItest.htm
In order to get the maxCPU behavior one needs to use the SRM, rather than directly launching the individual scripts.
E:\\SpectrumMill>millscripts\submitRequest.pl batchTagPara.pl millauto\mstag.name.params dataDir1 > msdataSM\dataDir1\mstag.CLItest.htm
One can also run an entire workflow via the SRM:
E:\\SpectrumMill>millscripts\workflow.pl workflow.name.tsv dataDir1 dataDir2 ... > msdataSM\dataDir1\workflow.CLItest.htm
More information is available on creation of Parameter Files (*.params). The simple name=value formatted
lines in a .params file is derived from the use of the PERL CGI package command: $queryCGI->save(\*PARAMSFILE)
Some of these PERL scripts (runXtractor.pl, batchTagPara.pl) launch C++ programs
that process batches of LC-MS/MS runs (xtractorAgilent.cgi, xtractorFinnigan.cgi) or batches of MS/MS spectra(mstagpara.cgi)
The batches are created from items located in a single directory. The C++ programs can receive their params via either CGI or CLI.
The PERL script (batchSherenga.pl) similarly processes batches of MS/MS spectra by running a Java program (sherenga.jar). Sherenga
receives parameters only by CLI. In max CPU mode, each PERL script is run once to create all the batches. Then the SRM directly
launches the corresponding C++ program (?via CLI or CGI?) or the Java program Sherenga (via CLI) for each batch. For MS/MS Search and
de novo Sequencing the SRM relies on the BatchTasks.txt and SherengaBatchTasks.txt files produced by the PERL scripts.
The SRM receives data directories by CLI and parameters by CGI. The SRM sends all parameters (including data directories) to the
PERL scripts by CGI, except for batchSherenga.pl (by CLI).
To Use the Workflows Form
The Workflows form is the entry point to execute and edit workflows. You can also view the parameters
associated with a particular parameter file. The following topics describe options available on the Workflows
form.
Data Directories
- Click the Select ... button to select one or more data directories. See
Selecting Data Directories. If you select multiple data directories,
make sure they are all of the same type (for example, all .raw
or all .d).
Workflow
- Execute - Click to process a workflow. Click this button after you have selected one
or more directories to process, and you have clicked a workflow in the Workflow list. When
you click the Execute button, each task in the selected workflow is submitted to the request queue
for each data folder. The tasks execute in order and in parallel, subject to data folder dependencies
and the available CPUs on the Spectrum Mill server. As each task is submitted, the request submission
information and links are displayed in the bottom frame of the form.
- Maximize CPUs: Mark the appropriate check box(es) if you want
Extraction, MS/MS Search, and/or de novo to take advantage of all
available CPUs. Otherwise, each will use only a single CPU so that the other CPUs are available for other
processes/users. Maximize CPUs applies to data within a
directory. Separate directories are always executed in parallel.
For MS/MS Search, and/or de novo Maximize CPUs is checked by default and should always improve performance.
However, for Extraction performance is only improved when a directory contains multiple files (LC-MS/MS runs).
Furthermore, the amount of system memory (RAM) may be a limiting factor for extraction of multiple files
in parallel. The SRM attempts to automatically limit the number of simultaneous Extractions based on file size, total system memory,
and number of processors. The automated limiting of simultaneous processes is both imperfect and conservative, because
prior to launching the Extractor the SRM is unable to determine whether a file has been acquired in profile or
centroid mode. The total amount of memory required, relative to file size, for Extraction is much less for a profile mode file.
For more information, see Multicore (Maximize CPUs) Data Extraction.
- Remove prior results (for extraction or first search) - Mark this check box to remove prior
extraction and MS/MS search results for the data folder(s) you selected. This will also remove
Spectrum Summary results.
- Edit Workflow - Click to display the Edit Workflow form, where you can edit a workflow,
or create and save a new workflow. See To Use the Edit Workflow Form.
- Refresh - Click to update the Workflow list when you (or other users) have created
new workflows, or have changed the tasks within a workflow.
- Workflow: Shows all available workflows. Click a workflow to see its tasks (in order
of execution) in the Tasks box to the right.
- Tasks: Shows the tasks in the workflow that you selected in the box to the left. Click a task
to see that task’s page in read-only mode in the bottom frame. This allows you to quickly confirm that
the parameters are appropriate, or to explore a particular workflow.
To Use the Edit Workflow Form
This form allows you to edit a workflow, or to create new workflows. To
create a new workflow, edit an existing one and save the changes to a
new name. See Chapter 3, Automating Workflows, of the Application
Guide to learn how to create new workflows.
To get to this form:
- In the Workflows form, click the name of the workflow you wish to edit.
- Click the Edit Workflow button.
- Verify that the Edit Workflow form shows the name of the workflow in the red title bar, and its
list of (ordered) tasks in the Workflow tasks list.
Workflow tasks are parameter files, which you create in each form.
The following describes options available on the Edit Workflow form:
Edit Workflow Tasks
- Available tasks: The list box initially shows all available tasks that have been defined.
To filter the list to show only a particular task type (for example, Extraction), click the down-arrow
and select the type of task you want to show.
- Refresh - Click to update the list of available tasks. This is necessary if you (or another
user) defines a new task in another browser window while the Edit Workflow window is open.
- To see a read-only display of the parameters for one of the Available tasks
or one of the Workflow tasks, simply click the task.
- Add-> To add a task to the list under Workflow tasks, click the task, then click
Add->. The program adds the task to the bottom of the workflow, but you can move it with the
Up and Down buttons.
- Workflow tasks: Displays the list of tasks in the workflow, in the order they are executed.
- Open - Click to display the Open Workflow dialog box, which lets you open a different
workflow. The title bar changes to indicate the new workflow, and its tasks are displayed in the Workflow
tasks list. See Open Workflow Dialog Box.
- Save As - Click to display the Save Workflow dialog box, which lets you save the workflow
with a new name or the same name. See Save Workflow Dialog Box.
- Up - To reorder the Workflow tasks by moving a task up, click the task, then click
Up.
- Down - To reorder the Workflow tasks by moving a task down, click the task, then click
Down.
- Remove - To remove a task from the Workflow tasks, click the task, then click Remove.
- Clear All - Click to remove all tasks from the workflow.
After you edit a workflow:
- Click the Save As button to save it.
- To see the changes in the Workflows form, click the Refresh button within that form.
To Use the Request Queue Viewer
The Request Queue Viewer shows a list of all tasks that are currently executing and those that are queued
for execution. It lists the tasks in the order they were queued. Because some tasks depend upon earlier tasks,
the tasks that are currently executing do not always appear at the top of the list.
- Remove - To remove a task from the queue, mark the checkbox under the ‘X’ image. Then
click Remove. When you remove tasks from the queue, the program still displays them in the
Completion Log, but it marks them as Aborted. If a task has begun executing, the Status
says Running, and you cannot select the task for deletion. If a
checkbox does not appear next to the task, you cannot stop it.
Notes:
- The Request Queue is not automatically updated. To refresh, click the Request Queue
button. Do not use the Refresh command that is built into Internet Explorer.
- The program assigns each task a Task Id, and displays it in the Request Queue viewer.
Under each Task Id is the word Monitor. Click Monitor to display the progress of
a task in a separate browser window. Monitor is only shown once a task
is running.
- If a task is dependent upon other tasks, they are listed in the Dependencies column. For example,
an Autovalidation task is dependent on an MS/MS Search task; the MS/MS Search must complete before Autovalidation
can execute.
- If you maximize CPUs, the program will create subtasks for each batch in
Extraction and MS/MS Search. The subtasks execute while the major task
is still queued. The Task Id will have a P (for
"parallel") appended to indicate that it is an extraction or search that is using the maximum available CPUs.
A parallel task's Status will indicate the progress for the
task as (#completed:#total).
The Completion Log will have two entries: one for the initial parallel
task request (with a "P" appended) that creates the batches to extract
or search as sub-tasks, and another that shows the completion status for each sub-task batch
extraction or search.
- The Request Queue shows "running" in green font for the task that is
currently executing.
- Task Types are shown for both the Request Queue and Completion Log Viewers as:
- extractorName of Instrument Vendor - extraction
- mstag - MS/MS search
- validate - autovalidation
- PPSummary - protein/peptide summary
- msfit - PMF search
- pmfsummary - PMF summary
- sherenga - Sherenga de novo sequencing
- sherengaReport - Sherenga de novo summary
- qualityMetrics - Quality Metrics & FDR
- archiveData - Archive Data
To Use the Completion Log Viewer
The Completion Log viewer shows a list of all tasks that have completed, with the most
recent shown at the top. This log includes all queued requests, whether you queued them interactively or via a
workflow. Two of the columns allow you to view additional information:
- Task Id - Click to display the saved results html file. This is particularly useful if the
task is a Protein/Peptide Summary task, because it shows the summary results.
- Data Set Parameters - Click to show a table of the parameters used for the task to the left.
You can view parameters for Data Extractor, MS/MS Search, and Autovalidation. Protein/Peptide Summary
tasks show no parameters.
To Use the Open Workflow Dialog Box
The Open Workflow dialog box allows you to select a workflow in the Edit Workflow form.
- Select Workflow: Shows all the available workflows. To open a workflow, click the name of
the workflow, then click the Open button.
- Workflow Tasks: Lists the tasks in the selected workflow. Note that in this form, you cannot
view the parameters for the tasks.
- Open - Click to open a workflow.
- Cancel - Click to stop without opening a workflow.
- Help - Click to display Help for the dialog box.
To Use the Save Workflow Dialog Box
The Save Workflow dialog box allows you to save a workflow that you have created in the Edit Workflow
form.
- Folder: Type or select the name of the folder where you want to save the workflow. Do not
use the forbidden characters (described in the dialog box) in the folder name. To create a new folder,
click the New folder icon.

- New folder icon - Opens the New Folder dialog box, which
allows you to create a new folder to store workflows. The folder is created under \SpectrumMill\millauto.
You may create only one level of folders in \SpectrumMill\millauto.
- Name - Type a name for the workflow, or click a name under Existing files.
- Existing files - Lists all the available workflows. To overwrite a workflow, click its name.
- Save - Click to save the workflow.
- Cancel - Click to stop without saving a workflow.
- Help - Click to display Help for the dialog box.
To Use the New Folder Dialog Box
The New Folder dialog box allows you to create a new folder where you save a workflow.
- New folder name: Type the name of the folder where you want to save the workflow. Do not
use the forbidden characters (described in the dialog box) in the folder name. The folder is created
under \SpectrumMill\millauto. You may create only one level of folders in \SpectrumMill\millauto.
- Existing folders - Lists the folders that already exist under \SpectrumMill\millauto.
- OK - Click to create the new folder.
- Cancel - Click to stop without creating a new folder.
- Help - Click to display Help for the dialog box.