Spectrum Mill MS/MS Search
Table of Contents
Introduction
The MS/MS Search module in Spectrum Mill automates the search of processed MS/MS spectra against
protein or DNA databases. The MS/MS Search algorithm uses intelligent parallelization to provide extremely fast
searches. It can operate in identity mode to find unmodified peptides or in variable modifications or homology
modes to look for mutations, post-translational modifications, and chemical modifications.
As you process data with Spectrum Mill, you may iterate through multiple rounds of database search
and results validation, with the goal of identifying as many spectra as possible. Spectrum Mill
provides a means to segregate search results that contain a valid interpretation of an MS/MS spectrum from those
that do not. Spectra that do not have validated matches can then be subjected to subsequent rounds of searches
(against larger databases or in variable mode, for example). Spectrum Mill retains a cumulative
list of validated matches that you can summarize at any time in the process.
What is a Fragment-Ion Tag?
A Fragment-Ion Tag can be obtained from an MS/MS spectrum and consists of three attributes:
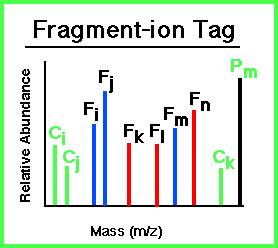 |
- A peptide precursor-ion mass: Pm.
- Masses of all sequence related fragment-ions from the peptide: Fi, Fj,
... The fragment ions need not all be of the same ion type. Supported
fragment-ion types are described below.
- Masses of composition ions which indicate the presence of particular amino acids in the peptide:
Ci, Cj, ... This information can be from immonium and related low
mass ions or high mass ions representing side-chain losses from the precursor ion.
|
Search Mode
Definitions
The following modes are available for MS/MS Search:
- Identity: The MS/MS spectra are compared to unmodified sequences from the database.
- Variable modifications: In addition to searching for identical matches, the search includes
the variable modifications you have selected. These may be post-translational modifications or other
variable modifications. Since they are considered as possible modifications, the search does not
force universal application of variable modifications. Peptides can have more than one variable modification,
within the limits of the user-specified Precursor mass shift. The search
does not currently support multiple types of variable modifications to the same type of amino acid.
For example, you cannot select both guanidination and carbamylation of lysine as variable modifications
within the same search.
- Homology - All mutations: In addition to searching for identical matches, the search includes
matches that are consistent with a single amino acid substitution. The substitution does not need to
be one that would result from a point mutation. If you have selected variable modifications for an amino
acid, they are also considered.
- Homology - Single base pair mutations: In addition to searching for identical matches,
the search includes matches that are consistent with a single amino acid substitution that would result
from a point mutation (single base substitution within a codon). If you have selected variable modifications
for an amino acid, they are also considered.
To search for an unknown or unexpected modification, select one of the homology modes described above, then
click Unassigned single mass gap. This search looks for an unexpected modification (a mass gap).
It is a way of doing an "error tolerant database search." Note that
you cannot simultaneously search variable modifications in this mode. This type
of search is best done when searching previous non-validated hits.
Tips:
Searches run faster and generate fewer false positives when fewer modifications are considered.
Your system administrator can define new mutation/substitution
matrices. Do not attempt to modify the existing homology modes.
Comparison of search modes
| |
Included in the search |
| |
Exact matches* |
Variable modifications** |
All single amino acid substitutions |
Only single amino acid substitutions that would result from a point mutation |
| Identity |
yes |
no |
no |
no |
| Variable modifications |
yes |
yes |
no |
no |
| Homology - All mutations
|
yes |
yes |
yes |
no |
Homology - All mutations- Unassigned single mass gap
|
yes |
no - only an unassigned modification |
no |
no |
| Homology - Single base pair mutations
|
yes |
yes |
no |
yes |
Homology - Single base pair mutations- Unassigned single mass gap
|
yes |
no - only an unassigned modification |
no |
no |
*Exact matches take into account fixed modifications, which are applied universally to their respective amino
acids. Exact matches also take into account any mix modifications that are applied during the search cycle.
**You must select the variable modifications you wish to search (Choose... button)
Search mode translator across Spectrum Mill versions
| Search mode and other settings |
| Identity |
| This mode is no longer supported. |
Variable modifications
Select the appropriate k, m, q, s, t, y combinations as variable modifications.
Set Precursor mass shift range as shown in the following table:
| Mode |
Precursor mass shift range |
| Homology Multi - mq |
-18 to 33 |
| Homology Multi - sty |
0 to 241 |
| Homology Multi - mqsty |
-18 to 257 |
| Homology Multi - mqst |
-18 to 257 |
| Homology Multi - mqy |
-18 to 177 |
| Homology Multi - kmqsty |
-18 to 257 |
| Homology Multi - kmq |
-18 to 101 |
|
Homology – All mutations
Select k, m, q, s, t, and y as variable modifications.
Set Precursor mass shift to +/- 81 |
| Homology – Single base pair mutations |
Homology Mode
In order to match one's data to a sequence in the database that is not identical to the peptide used to generate
the MS/MS spectrum, MS/MS Search must be used in homology mode. This enables matching for peptides with a mutation,
cross-species substitution, sequence polymorphism, or error in the database. Homology mode works based on three
concepts:
- Allow precursor mass to be shifted from the precursor mass of sequences in the database.
- Consider each ion independently rather than examining relationships between ions.
- When a precursor ion undergoes fragmentation, at least two pieces are formed (a fragment-ion and a neutral).
While only the mass of the ionized fragment is measured, the mass of the neutral piece is easily calculated
as precursor mass - fragment ion mass. If the peptide matches a database sequence exactly, then the masses
of BOTH the fragment-ion and the neutral will match. If there is a single sequence difference, then the mass
of either the fragment-ion or the neutral will match, but NOT BOTH. If there are two sequence differences,
fragmentation at any sites located BETWEEN the two mismatched sites will result in NEITHER the fragment-ion
nor the neutral matching.
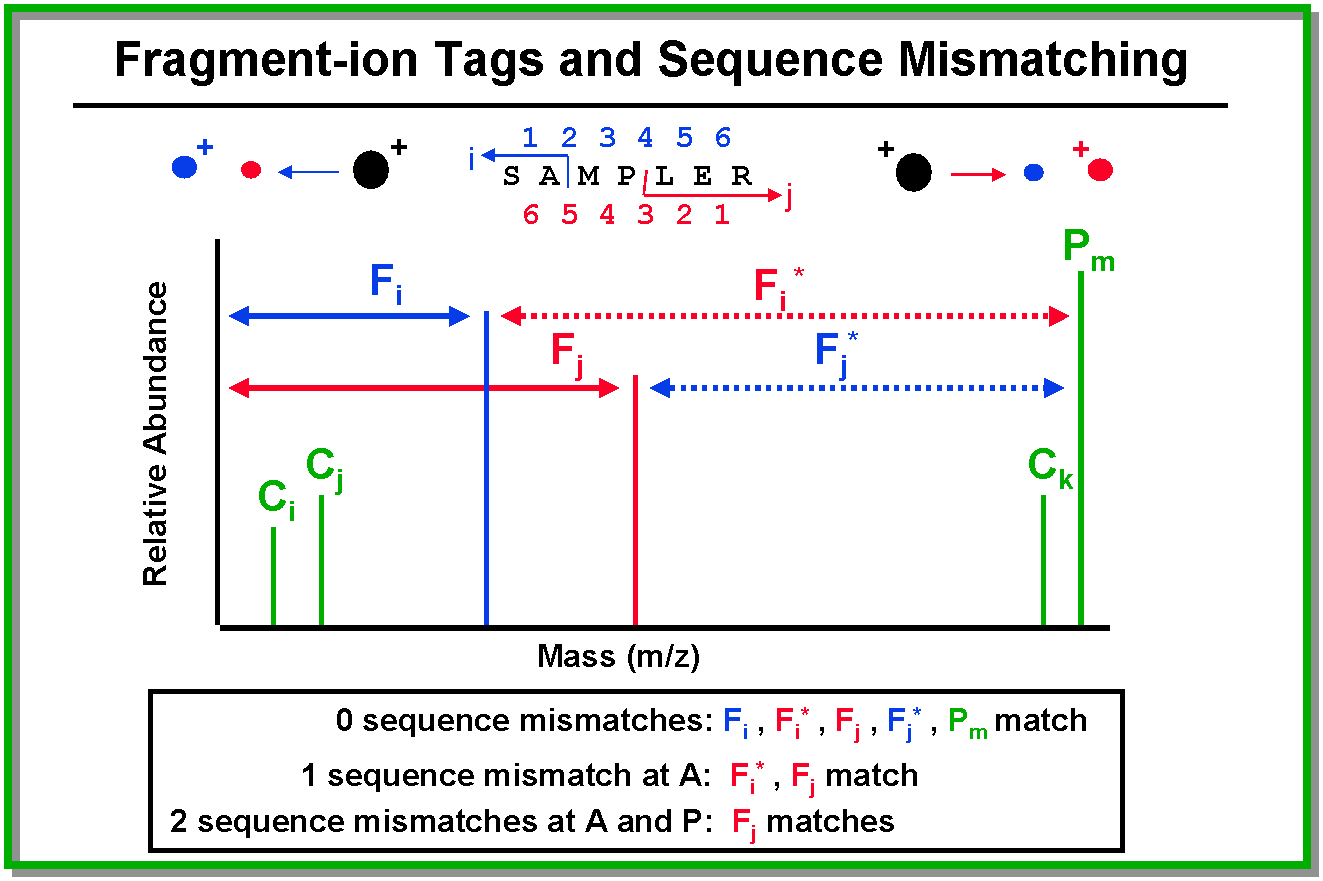
Matching sequences are filtered through a mutation matrix to try to find
a single amino acid (AA) substitution that would transform the calculated mass of the database sequence to the
experimentally determined mass. The output displays the necessary substitution and the corresponding sequence
consistent with the experimental peptide mass data (not the sequence present in the database).
Precursor Mass Shift
MS/MS Search only considers database sequences with calculated precursor masses which pass through a precursor
mass filter. In Identity Mode, the filter is determined by the specified precursor mass +/- the precursor mass
tolerance. In Homology mode this is determined by the specified precursor mass
and the precursor mass shift. You should NOT attempt to accomplish this by using a wider precursor mass tolerance.
Use a precursor mass tolerance consistent with the accuracy to which the precursor mass is measured. The default
value of +/- 130 allows for the largest possible precursor mass shift associated with a mutation among the 20
standard amino acids and phosphorylation. All database sequences with a calculated precursor mass + / - 130 Da
of the specified precursor mass would thus be considered. This means a large increase in the number of sequences
considered, and hence increases the potential for false-positives. The +/= and -/= features allow you to specify
an anticipated precursor mass shift value and reduce the number of sequences considered in a search. For example,
suppose you expect a phosphorylated peptide; specifying a precursor mass shift of +/= 80 would allow matches to
database sequences that exactly match the precursor mass or database sequences that would match the specified
precursor mass if 80 Da were added.
The default precursor mass shift range for the Variable modifications search mode is -18 to 177.
You can change this setting to encompass the number and type of modifications you expect for your sample.
To summarize, the shift can be set in four different forms, all of which show only homologous matches, thus
excluding identity mode matches:
- +/- (wide range) - allows matching of a query spectrum to all library spectra spanning the range
of the Precursor mass shift
- =/+/- allows a query spectrum to match a library spectrum only if the query spectrum's precursor
MH+ is shifted either higher or lower by the specified mass. (The program automatically takes
into account precursor charge.)
- +/= (specified shift up) - allows a query spectrum to match a library spectrum only if the query
spectrum's precursor MH+ is shifted higher by the specified mass. (The program automatically
takes into account precursor charge.)
- -/= (specified shift down) - allows a query spectrum to match a library spectrum only if the query
spectrum's precursor MH+ is shifted lower by the specified mass. (The program automatically
takes into account precursor charge.)
Note that the +/- will compare many more spectra so it will take longer to run, and the run time will be proportional
to the magnitude of the Precursor mass shift.
Ranking / Scoring of Results
The explanations below are for a simple score calculation. With version B.04.00
and later, you also have the option to select Discriminant Scoring. See Discriminant scoring to learn more about the "figures of merit" that comprise the discriminant score and search mode to learn about the discriminant scoring options from which you can choose.
Scoring Scheme
Following peak detection, the MS/MS Search algorithm attempts to match
every ion present in an MS/MS spectrum to an ion type consistent with fragmentation of a peptide sequence from
a database. The scoring system is information-content oriented and based on the following general principles:
- Peak Intensity - If a peak is "real" and explainable, intensity doesn’t matter. Very intense
unexplained peaks suggest poor interpretation.
- Fragment Ion Types - Secondary ion types have redundant information content.
- Peptide Fragmentation - Sequence completeness depends on backbone cleavages (e.g., b or y
ions).
- Peptide Length - Longer peptides yield more ions and thus the spectra contain more information.
- Protein Identity - One good peptide MS/MS is better than three mediocre ones.
- Proton Mobility - This scoring refinement for ion trap MS/MS data is based on the likelihood of
fragmentation at a particular location. When you mark the check box for proton mobility scoring in MS/MS
Search, the software takes into account relative proton mobility based on peptide charge state and amino
acid composition. It also considers the position of an amino acid in a peptide and the peptide length.
For details, see:
Kapp, E. A.; Schutz, F.; Reid, G. E.; Eddes, J. S.; Moritz, R. L.; O'Hair, R. A. J.; Speed, T. P.; Simpson,
R. J.; "Mining a Tandem Mass Spectrometry Database To Determine the Trends and Global Factors Influencing
Peptide Fragmentation;" Anal. Chem.; 2003; 75(22); 6251-6264. DOI:
10.1021/ac034616t
Do not use proton mobility scoring when peptides are modified in a way that significantly alters the
way the peptide fragments. You can use proton mobility scoring for 14N/15N,
SILAC, and 16O/18O because these isotopic labels do not change the structure of the
peptides and thus do not change the way these peptides fragment. However, the iTRAQ labels and the lys-imidazole
labels do change the way a peptide fragments and thus proton mobility scoring is not advised for these.
It is also not advised for guanidination or phosphorylation.
The scoring scheme is intended to facilitate review/filtering of large numbers of spectra (100's - 1000's)
that enables segregating valid from false-positive interpretations. Note that probability values associated with
the number of proteins/peptides in the database are NOT used. Thus the score for a spectrum against a particular
candidate sequence will always be the same, as the information content of the spectrum is database-independent.
MS/MS Search scoring has four particular attributes. Only the first two are used for post-search review/filtering
purposes within the Protein/Peptide Summary portion of Spectrum Mill.
- Score - The software calculates the MS/MS Search score as described in the following bullets
and diagram. To see details about the ions that contribute to any given score, click the link under the
Filename header in Protein/Peptide Summary.
- Bonus points for each peak assigned to an allowed fragment ion type for a candidate
peptide sequence. Bonus values of particular ion types are instrument-dependent.
example: (ESI-ION-TRAP b/y: 1, a: 0.25) Marker ions can also
contribute to bonus scoring. - Penalty points for each unassigned peak. Penalty value is based on peak height, -
(peak height / height of tallest peak).
example: if an unassigned peak is 50% the height of the tallest peak in the peak-detected
spectrum, then its penalty value is 0.5, while an unassigned peak that is 10% the height of
the tallest peak has a penalty value of only 0.1.
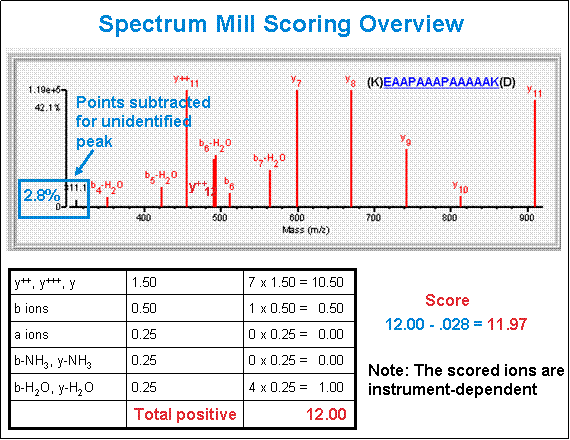
- SPI - Scored Peak Intensity - From peaks remaining after peak detection, the percentage of
total intensity which is assigned to particular ion types. Scored Peak Intensities lower than 70% suggest
a poor interpretation, or presence of fragment ion types unknown to MS/MS Search.
- BCS - Backbone Cleavage Score - Number of amino acids cleaved between either b or y ions.
- Unmatched ions - # of unassigned ions / # of ions remaining after peak detection.
Guidelines for correlating peptide score with the correct interpretation and the extent of
peptide fragmentation:
The correct cut-off score is dependent on the sample complexity and type. These values should be used as guidelines only.
Q-TOF
- Score > 15 Outstanding, well-assigned, thorough fragmentation
- Score > 9 Good
- Score > 5 Mixed Quality
- Score < 5 Generally poor with less fragmentation
- SPI < 60% Generally poor with significant unassigned ion current
Ion trap
- Score > 15 Outstanding, well-assigned, thorough fragmentation
- Score > 10 Good
- Score > 5 Mediocre, relatively few fragment ions produced, look for mostly b's or
mostly y's.
- Score < 5 Poor, little to no fragmentation
- SPI < 70% Poor, significant unassigned ion current
Fragmentation and Sequence Information Content
The figure below illustrates some of the diversity of quality and information content in MS/MS spectra.
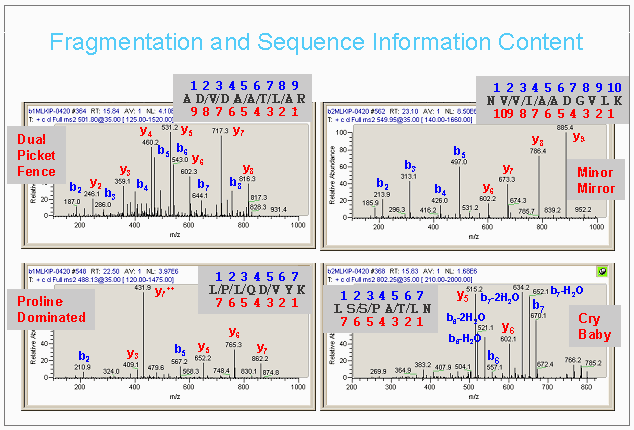
If scores for a single spectrum against multiple candidate sequence are identical, the results are then sorted
so that if multiple sequences are matched, more likely sequences are listed higher in the list and sorted on the
following basis:
- In Variable modifications mode and in Homology mode, matches
to database sequences identical to their appearance in the database are listed higher.
- In Variable modifications mode and in Homology mode, matches
with known modifications are listed higher.
- Sequences matching with the least number of unmatched ions are listed higher.
- Among equivalent matches, the results are sorted in order of increasing precursor mass.
- Among equivalent matches, the results are sorted in order of increasing index number.
Note that the last two sorts do NOT imply a BETTER ranking, even though one match will be listed
higher than another, but are merely intended to provide some organization to the listing and to aid the user in
viewing the results.
Discriminant Scoring
Discriminant
scoring was new with Spectrum Mill version B.04.00. If you enable
discriminant scoring on the MS/MS Search page, the discriminant
score is calculated in addition to the score, and you can select to use
the discriminant score for Autovalidation.
With Spectrum Mill B.05.00,
the default Discriminant mode is Off rather than Score
only. Update any workflow parameter files to use the new
settings.
The discriminant score is a combination of
several figures of merit into a single score, which helps discriminate
between real positives and false positives more successfully than score
alone. On average, the number of ID's with 1% and 5% false
discovery rates increases by 10% using a discriminant score over score
alone. That is, it will improve the Global and Local Peptide FDR
autovalidation because it includes additional factors other than the
score.
To use discriminant scoring you must either use an already validated
set of coefficients for the figures of merit for Agilent instruments or
create your own set of coefficients, which you can do in the Tool
Belt. They require a carefully validated data set that is typical
of the type to be used for future searches. The data set also
needs to be sufficiently large to generate reasonable coefficients.
These are the figures of merit from database
search scores included in the discriminant score:
- Score
- Delta rank1-rank2 score
- Backbone cleavage score
- Number of complementary fragments
- Maximum matched sequence tag length (b,y,c, or z ions)
- SPI%
- Percentage of matched ions (e.g., 17/25 peaks matched = 68%)
- Standard deviation of fragment mass errors/fragment tolerance
- Charge state
Instrument / Fragment-Ion Types
Selecting an instrument triggers the configuration of MS/MS Search scoring and
peak detection parameters designed particularly for the type and extent
of peptide fragmentation observed on that instrument. The particular parameters can be edited or
new instruments added by editing the files: msparams_mill/instrument.txt,
and millhtml/SM_js/instrument.js
E:\SpectrumMill\msparams_mill\instrument.txt
E:\SpectrumMill\millhtml\SM_js\instrument.js
If you add a new instrument type, be sure to set the parameters in instrument.txt in a way that
is appropriate for the data you export from that instrument. For example, if deisotoping is accomplished by
the instrument data system, set bypassDeIsotoping = 1 in instrument.txt to avoid repeating deisotoping
in Spectrum Mill.
Examples of supported instrument configurations are shown in the three tables below. Agilent ESI instruments
are described in the first table, while other ESI instruments are described in the second. MALDI instruments are
described in the third table. For additional supported configurations
and the latest updated settings, see
E:\SpectrumMill\msparams_mill\instrument.txt.
Table 1. Examples of supported configurations for Agilent instruments
| Feature |
Description |
ESI-ION-TRAP-Agilent |
ESI-ION-TRAP-Agilent-ETD |
ESI-QTOF-Agilent |
| nh3_loss |
NH3 loss residues |
R, K, Q |
R, K, Q |
R, K, Q, N |
| h2o_loss |
H2O loss residues |
S, T, E, D |
S, T, E, D |
S, T, E, D |
| pos_charge |
charge-bearing residues |
R, H, K, N, Q |
R, H, K, N, Q |
R, H, K, N, Q |
| instrument charges certain |
fragment charges certain (allows ambiguity in charge) |
no |
no |
if determined |
| min_fragment_mass |
discards peaks below
impacts immonium ion detection capability |
105 |
105 |
58 |
| max_internal_ion_mass |
impacts search speed
if internal ions allowed |
N/A |
N/A |
750 |
| minSignalNoiseRatio |
threshold for peak detection |
8 |
0 |
8 |
| minSignalNoiseRatioPMF |
threshold for peak detection in MS-only mode |
|
5 |
15 |
| peakLimitCount |
max # of detected peaks to use for interpretation |
25 |
25 |
25 |
| peakBinningTolerance |
used for centroiding in Data Extractor - expected peak width in amu |
N/A |
N/A |
0.1 |
| bypassDeIsotoping |
skip de-isotoping |
no |
no |
no |
| bypassSignalNoiseThresholding |
skip S/N thresholding |
no |
no |
yes |
| composition_bonus_scoring |
MALDI equivalent to proton mobility scoring, where bonuses are applied only to fragments on the
N-terminal side of aspartic or glutamic acid and the C-terminal side of proline, scaled based on the
relative intensity of the fragment. It does not give a bonus to any other amino acid. |
no |
no |
no |
| merge_num_peaks |
For similarity merging of MS/MS spectra, the number of peaks that match between the two spectra
must be greater than or equal to merge_num_peaks, which is a number between 0 and 50. The similarity
merging takes the top 50 peaks from both spectra and compares them. All instruments that generate
MS/MS data use the default merge_num_peaks = 25, but if you add an entry to instrument.txt,
your entry overrides the default. The format is merge_num_peaks, followed by a tab, followed
by the value. |
25 (default) |
25 (default) |
5 |
| merge_SPI |
For similarity merging of MS/MS spectra, the percentage of the total intensity of the top 50 spectral
peaks that is matched from spectrum A to spectrum B and from spectrum B to spectrum A must be greater
than or equal to merge_SPI, which is a number between 0 and 100. All instruments that generate
MS/MS data use the defaults of merge_SPI = 70, but if you add an entry to instrument.txt,
your entry overrides the defaults. The format is merge_SPI, followed by a tab, followed
by the value. |
70 (default) |
70 (default) |
50 |
| minValidMSMSScore |
Scores lower than this setting are ignored during search. Lower values allow smaller peptides
to be kept as possible hits, at the risk of adding more false hits. Note that this setting also affects
reverse scores. |
3 (default) |
0 |
3 (default) |
| minMSMSScoreForOutputFile |
If the score is lower than this setting, the spo file is not generated. This helps limit “file
clutter”. For small peptides, use a smaller number. |
3 |
0 |
3 |
| Ion type |
Restrictions |
ESI-ION-TRAP-Agilent
Score |
ESI-ION-TRAP-Agilent-ETD |
ESI-QTOF-Agilent
Score |
| a |
none |
0.25 |
N/A |
0.50 |
| b, y |
none |
1.00 |
N/A, 0.25 |
0.5, 1.5 |
| a-NH3 |
contains NH3 loss residue |
N/A |
N/A |
N/A |
| b-NH3, y-NH3 |
contains NH3 loss residue |
0.50 |
N/A |
0.25, 0.5 |
| b-H2O, y-H2O |
contains H2O loss residue |
0.50 |
N/A |
0.25, 0.5 |
| b+H2O |
ion contains charge bearing residue
only bn-1, bn-2 ( length n) |
1.00 |
N/A |
1.00 |
| d(H) |
AA is A,C,D,E,K,M,N,R,Q, or S |
N/A |
N/A |
N/A |
| d(CH3) |
AA is I,T,or V |
N/A |
N/A |
N/A |
| w(H) |
AA is A,C,D,E,K,M,N,R,Q, or S |
N/A |
N/A |
N/A |
| w(CH3) |
AA is I,T,or V |
N/A |
N/A |
N/A |
| b++, b+++, y++, y+++ |
fragment charges not certain
precursor charge > 2 (++), > 3 (+++)
ion contains sufficient charge bearing residues |
1.00 |
N/A, N/A, 0.25, 0.25 |
0.5, 0.5, 1.5, 1.5 |
| b++-H2O, y++-H2O |
fragment charges not certain
precursor charge > 2 (++)
ion contains > 1 charge bearing residue
contains H2O loss residue
corresponding b++, y++ present |
0.50 |
N/A |
0.25, 0.5 |
| a-H3PO4 |
ion contains phosphorylated S, T, Y
automatically turned on in homology mode
following detection of M-H3PO4 |
N/A |
N/A |
N/A |
| b-H3PO4, y-H3PO4 |
ion contains phosphorylated S, T, Y
automatically turned on in homology mode
following detection of M-H3PO4 |
0.25 |
N/A |
0.50 |
| b-SOCH4, y-SOCH4 |
ion contains oxidized M
automatically turned on in homology mode
following detection of M-SOCH4 |
0.25 |
N/A |
N/A |
| internal b |
< max_internal_ion_mass |
N/A |
N/A |
0.75 |
| internal a |
< max_internal_ion_mass, internal b present |
N/A |
N/A |
0.50 |
| internal b-H2O |
< max_internal_ion_mass, internal b present
ion contains H2O loss residue |
N/A |
N/A |
0.50 |
| internal b-NH3 |
< max_internal_ion_mass, ion contains R |
N/A |
N/A |
0.50 |
| N-term ladder |
removal of N-term residues (y equiv.) |
N/A |
N/A |
N/A |
| C-term ladder |
removal of C term residues (b+H2Oequiv.) |
N/A |
N/A |
N/A |
| c |
cannot cleave at proline |
N/A |
1.00 |
N/A |
| c++, c+++ |
cannot cleave at proline |
N/A |
1.00 |
N/A |
| z· |
cannot cleave at proline |
N/A |
1.00 |
N/A |
| z·++, z·+++ |
cannot cleave at proline |
N/A |
1.00 |
N/A |
| c·, c·++, c·+++ |
cannot cleave at proline |
N/A |
0.25 |
N/A |
| z··, z··++, z··+++ |
cannot cleave at proline |
N/A |
0.25 |
N/A |
*N/A = not applicable
Table 2. Examples of ESI configurations
| Feature |
Description |
ESI-ION-TRAP |
ESI-LINEAR-ION-TRAP |
ESI-QTRAP |
ESI-QSTAR |
ESI-QTOF |
| nh3_loss |
NH3 loss residues |
R, K, Q |
R, K, Q |
R, K, Q, N |
R, K, Q, N |
R, K, Q, N |
| h2o_loss |
H2O loss residues |
S, T, E, D |
S, T, E, D |
S, T, E, D |
S, T, E, D |
S, T, E, D |
| pos_charge |
charge-bearing residues |
R, H, K, N, Q |
R, H, K, N, Q |
R, H, K, N |
R, H, K, N, Q |
R, H, K, N, Q |
| instrument charges certain |
fragment charges certain (allows ambiguity in charge) |
no |
no |
no |
if determined |
if determined |
| min_fragment_mass |
discards peaks below
impacts immonium ion detection capability |
105 |
105 |
105 |
105 |
105 |
| max_internal_ion_mass |
impacts search speed
if internal ions allowed |
N/A |
N/A |
750 |
750 |
750 |
| localSignalNoiseRatio |
Signal-to-noise is calculated in local windows 100 m/z wide above the precursor m/z,
and 70 m/z wide below. The window width is increased in integer multiples if there are less
than 30 data points in the window above the precursor, or less than 20 data points in the window below
the precursor. |
no |
yes |
no |
no |
no |
| minSignalNoiseRatio |
threshold for peak detection |
8 |
8 |
8 |
8 |
8 |
| minSignalNoiseRatioPMF |
threshold for peak detection in MS-only mode |
|
|
|
15 |
|
| peakLimitCount |
max # of detected peaks to use for interpretation |
25 |
25 |
25 |
25 |
25 |
| peakBinningTolerance |
used for centroiding in Data Extractor - expected peak width in amu |
N/A |
N/A |
0.95 |
0.3 |
N/A |
| bypassDeIsotoping |
skip de-isotoping |
no |
no |
no |
no |
no |
| bypassSignalNoiseThresholding |
skip S/N thresholding |
no |
no |
no |
no |
no |
| composition_bonus_scoring |
MALDI equivalent to proton mobility scoring, where bonuses are applied only to fragments on the
N-terminal side of aspartic or glutamic acid and the C-terminal side of proline, scaled based on the
relative intensity of the fragment. It does not give a bonus to any other amino acid. |
no |
no |
no |
no |
no |
| merge_num_peaks |
For similarity merging of MS/MS spectra, the number of peaks that match between the two spectra
must be greater than or equal to merge_num_peaks, which is a number between 0 and 50. The similarity
merging takes the top 50 peaks from both spectra and compares them. All instruments that generate
MS/MS data use the default merge_num_peaks = 25, but if you add an entry to instrument.txt,
your entry overrides the default. The format is merge_num_peaks, followed by a tab, followed
by the value. |
25 (default) |
25 (default) |
25 (default) |
25 (default) |
25 (default) |
| merge_SPI |
For similarity merging of MS/MS spectra, the percentage of the total intensity of the top 50 spectral
peaks that is matched from spectrum A to spectrum B and from spectrum B to spectrum A must be greater
than or equal to merge_SPI, which is a number between 0 and 100. All instruments that generate
MS/MS data use the defaults of merge_SPI = 70, but if you add an entry to instrument.txt,
your entry overrides the defaults. The format is merge_SPI, followed by a tab, followed
by the value. |
70 (default) |
70 (default) |
70 (default) |
70 (default) |
70 (default) |
| Ion type |
Restrictions |
ESI-ION-TRAP
Score |
ESI-LINEAR-ION-TRAP
Score |
ESI-QTRAP
Score |
ESI-QSTAR
Score |
ESI-QTOF
Score |
| a |
none |
0.25 |
0.25 |
0.25 |
0.50 |
0.50 |
| b, y |
none |
1.00 |
1.00 |
1.00 |
1.00 |
1.00 |
| a-NH3 |
contains NH3 loss residue |
N/A* |
N/A |
N/A |
N/A |
N/A |
| b-NH3, y-NH3 |
contains NH3 loss residue |
0.50 |
0.50 |
0.25 |
0.25 |
0.25 |
| b-H2O, y-H2O |
contains H2O loss residue |
0.50 |
0.50 |
0.25 |
0.25 |
0.25 |
| b+H2O |
ion contains charge bearing residue
only bn-1, bn-2 ( length n) |
1.00 |
1.00 |
1.00 |
1.00 |
1.00 |
| d(H) |
AA is A,C,D,E,K,M,N,R,Q, or S |
N/A |
N/A |
N/A |
N/A |
N/A |
| d(CH3) |
AA is I,T,or V |
N/A |
N/A |
N/A |
N/A |
N/A |
| w(H) |
AA is A,C,D,E,K,M,N,R,Q, or S |
N/A |
N/A |
N/A |
N/A |
N/A |
| w(CH3) |
AA is I,T,or V |
N/A |
N/A |
N/A |
N/A |
N/A |
| b++, b+++, y++, y+++ |
fragment charges not certain
precursor charge > 2 (++), > 3 (+++)
ion contains sufficient charge bearing residues |
1.00 |
1.00 |
1.00 |
1.00 |
1.00 |
| b++-H2O, y++-H2O |
fragment charges not certain
precursor charge > 2 (++)
ion contains > 1 charge bearing residue
contains H2O loss residue
corresponding b++, y++ present |
0.50 |
0.50 |
0.25 |
0.25 |
0.25 |
| a-H3PO4 |
ion contains phosphorylated S, T, Y
automatically turned on in homology mode
following detection of M-H3PO4 |
N/A |
N/A |
N/A |
N/A |
N/A |
| b-H3PO4, y-H3PO4 |
ion contains phosphorylated S, T, Y
automatically turned on in homology mode
following detection of M-H3PO4 |
0.25 |
0.25 |
0.25 |
0.50 |
0.50 |
| b-SOCH4, y-SOCH4 |
ion contains oxidized M
automatically turned on in homology mode
following detection of M-SOCH4 |
0.25 |
0.25 |
0.25 |
0.25 |
N/A |
| internal b |
< max_internal_ion_mass |
N/A |
N/A |
0.75 |
0.75 |
0.75 |
| internal a |
< max_internal_ion_mass, internal b present |
N/A |
N/A |
0.25 |
0.50 |
0.50 |
| internal b-H2O |
< max_internal_ion_mass, internal b present
ion contains H2O loss residue |
N/A |
N/A |
N/A |
0.50 |
0.50 |
| internal b-NH3 |
< max_internal_ion_mass, ion contains R |
N/A |
N/A |
N/A |
0.50 |
0.50 |
| N-term ladder |
removal of N-term residues (y equiv.) |
N/A |
N/A |
N/A |
N/A |
N/A |
| C-term ladder |
removal of C term residues (b+H2Oequiv.) |
N/A |
N/A |
N/A |
N/A |
N/A |
*N/A = not applicable
Table 3. Examples of MALDI configurations
| Feature |
Description |
MALDI-ION-TRAP |
MALDI-TOF-TOF |
MALDI-TOF-TOF-DB |
MALDI-QTOF |
MALDI-QSTAR |
| nh3_loss |
NH3 loss residues |
R, K, Q |
R, K, Q |
R, K, Q |
R, K, Q |
R, K, Q |
| h2o_loss |
H2O loss residues |
S, T |
S, T |
S, T |
S, T |
S, T |
| pos_charge |
charge-bearing residues |
R, H, K |
R, H, K |
R, H, K |
R, H, K |
R, H, K |
| instrument charges certain |
fragment charges certain (allows ambiguity in charge) |
if determined |
if determined |
yes |
if determined |
if determined |
| min_fragment_mass |
discards peaks below
impacts immonium ion detection capability |
105 |
58 |
58 |
58 |
58 |
| max_internal_ion_mass |
impacts search speed
if internal ions allowed |
750 |
750 |
750 |
750 |
750 |
| minSignalNoiseRatio |
threshold for peak detection |
5 |
20 |
20 |
8 |
8 |
| minSignalNoiseRatioPMF |
threshold for peak detection in MS-only mode |
15 |
|
|
|
15 |
| peakLimitCount |
max # of detected peaks to use for interpretation |
25 |
25 |
25 |
25 |
25 |
| peakBinningTolerance |
used for centroiding in Data Extractor - expected peak width in amu |
N/A |
N/A |
N/A |
N/A |
0.6 |
| bypassDeIsotoping |
skip de-isotoping |
no |
yes |
yes |
no |
no |
| bypassSignalNoiseThresholding |
skip S/N thresholding |
no |
yes |
yes |
no |
no |
| composition_bonus_scoring |
MALDI equivalent to proton mobility scoring, where bonuses are applied only to fragments on the
N-terminal side of aspartic or glutamic acid and the C-terminal side of proline, scaled based on the
relative intensity of the fragment. It does not give a bonus to any other amino acid. |
yes |
yes |
yes |
yes |
yes |
| Ion type |
Restrictions |
MALDI-ION-TRAP
Score |
MALDI-TOF-TOF
Score |
MALDI-TOF-TOF-DB
Score |
MALDI-QTOF
Score |
MALDI-QSTAR
Score |
| a |
none |
0.50 |
0.50 |
0.50 |
0.50 |
0.50 |
| b, y |
none |
1.00 |
1.00 |
1.00 |
1.00 |
1.00 |
| a-NH3 |
contains NH3 loss residue |
N/A |
N/A |
N/A |
N/A |
N/A |
| b-NH3, y-NH3 |
contains NH3 loss residue |
0.50 |
0.50 |
0.50 |
0.50 |
0.50 |
| b-H2O, y-H2O |
contains H2O loss residue |
0.50 |
0.50 |
0.50 |
0.50 |
0.50 |
| b+H2O |
ion contains charge bearing residue
only bn-1, bn-2 ( length n) |
1.00 |
1.00 |
1.00 |
1.00 |
1.00 |
| d(H) |
AA is A,C,D,E,K,M,N,R,Q, or S |
N/A |
0.25 |
0.25 |
N/A |
N/A |
| d(CH3) |
AA is I,T,or V |
N/A |
0.50 |
0.50 |
N/A |
N/A |
| w(H) |
AA is A,C,D,E,K,M,N,R,Q, or S |
N/A |
0.25 |
0.25 |
N/A |
N/A |
| w(CH3) |
AA is I,T,or V |
N/A |
0.50 |
0.50 |
N/A |
N/A |
| b++, b+++, y++, y+++ |
fragment charges not certain
precursor charge > 2 (++), > 3 (+++)
ion contains sufficient charge bearing residues |
N/A |
N/A |
N/A |
N/A |
N/A |
| b++-H2O, y++-H2O |
fragment charges not certain
precursor charge > 2 (++)
ion contains > 1 charge bearing residue
contains H2O loss residue
corresponding b++, y++ present |
N/A |
N/A |
N/A |
N/A |
N/A |
| a-H3PO4 |
ion contains phosphorylated S, T, Y
automatically turned on in homology mode
following detection of M-H3PO4 |
N/A |
N/A |
N/A |
N/A |
N/A |
| b-H3PO4, y-H3PO4 |
ion contains phosphorylated S, T, Y
automatically turned on in homology mode
following detection of M-H3PO4 |
0.50 |
0.50 |
0.50 |
0.50 |
0.50 |
| b-SOCH4, y-SOCH4 |
ion contains oxidized M
automatically turned on in homology mode
following detection of M-SOCH4 |
N/A |
N/A |
N/A |
N/A |
N/A |
| internal b |
< max_internal_ion_mass |
0.75 |
0.75 |
0.75 |
0.75 |
0.75 |
| internal a |
< max_internal_ion_mass, internal b present |
0.50 |
0.50 |
0.50 |
0.50 |
0.50 |
| internal b-H2O |
< max_internal_ion_mass, internal b present
ion contains H2O loss residue |
0.50 |
0.50 |
0.50 |
0.50 |
0.50 |
| internal b-NH3 |
< max_internal_ion_mass, ion contains R |
0.50 |
0.50 |
0.50 |
0.50 |
0.50 |
| N-term ladder |
removal of N-term residues (y equiv.) |
N/A |
N/A |
N/A |
N/A |
N/A |
| C-term ladder |
removal of C term residues (b+H2Oequiv.) |
N/A |
N/A |
N/A |
N/A |
N/A |
*N/A = not applicable
Selecting Thermo Fisher Scientific Instruments
If you have a Thermo Fisher Scientific Orbitrap or LTQ FT, select your instrument based on where the MS/MS
occurs.
- If you have an Orbitrap:
- If MS/MS occurs in the LTQ, select the appropriate linear ion trap instrument and change
the Precursor mass tolerance to 0.05 Da (note that the units must be Daltons).
- If MS/MS occurs in the Orbitrap, select the appropriate Orbitrap instrument and keep
the defaults (in ppm).
- If you have a standard LTQ FT, select the appropriate linear ion trap instrument because the MS/MS
occurs in the LTQ. (Only precursor ion scans occur in the FT.) Set the Precursor mass tolerance
to 0.05 Da.
- If you have an LTQ FT Ultra, and MS/MS occurs in the FT, then select ESI Orbitrap.
Fragmentation modes and location
With
version B.04.00 a list of fragmentation modes is now available.
The list does away with the need for the "MIX" Instrument types
available in previous versions. Select an Instrument, then a fragmentation mode.
- CID - collision-induced dissociation in the LTQ
- ETD - electron transfer dissociation in the LTQ
- HCD - isolate precursor ions in the LTQ, dissociate in the C-trap, scan out the fragment ions and
detect in the Orbitrap
- PQD - pulsed-q dissociation - isolate precursor ions and dissociate in the LTQ, but without the low-mass
cutoff that you see with CID spectra
Multiply-Charged Ions
When data is of sufficient resolution that charge state can be determined from the isotope distribution,
and the software designates MS/MS Search instrument configuration as "fragment charges certain," then masses
are converted to charge 1 inside MS/MS Search prior to interpretation. However, the charge state is still used
to evaluate matching sequences to check that they contain a sufficient number of basic residues to support the
charge. Further, in the output, the labels distinguish whether the ion type used inside MS/MS Search was of the
converted to charge 1 high res variety (y+2) or of the ambiguous low res variety (y++).
Immonium Ions / Compositional Marker Ions
Marker ions represent peaks that indicate amino acid composition, but do not indicate sequence. The table
below describes the allowed amino acid composition marker ions. In general, the scores correspond to the rarity
of the amino acids as described by the number of codons coding for the amino acids that can produce the ion.
| Mass |
Composition |
Score |
Additional Feature / Constraint |
| 60 |
S |
2/6 |
|
| 70 |
PR |
2/10 |
|
| 72 |
V |
2/4 |
|
| 73 |
R |
2/6 |
|
| 86 |
IL |
2/9 |
|
| 88 |
D |
1 |
|
| 101 |
KQ |
2/4 |
|
| 102 |
E |
1 |
|
| 110 |
H |
1 |
|
| 112 |
R |
2/6 |
|
| 120 |
F |
1 |
|
| 129 |
KRQ |
2/10 |
|
| 136 |
Y |
1 |
|
| 159 |
W |
2 |
|
| (M+zH-H3PO4)+z |
sty |
2 |
variable mode required with those modifications selected
automatically turns on ion types b-H3PO4, y-H3PO4 |
(M+zH-284.2)+z
(M+zH-403.3)+z
(M+zH-477.3)+z |
C |
2 |
ICAT-D0 |
(M+zH-288.2)+z
(M+zH-411.3)+z
(M+zH-485.3)+z |
C |
2 |
ICAT-D8 |
(M+zH-270.2)+z
(M+zH-375.1)+z
(M+zH-449.3)+z |
C |
2 |
Acetyl-PEO-Biotin |
Note that the file msparams_mill\smconfig.xml defines additional marker ions and their scoring for
a large number of amino acid modifications. The scoring is invoked when the fixed and variable modifications are
selected for the search. System administrators can add custom modifications,
along with their marker ions.
For scoring purposes, one can not make a yes/no distinction between marker ions and peaks that are isobaric
with marker ions. So for scoring purposes, Spectrum Mill shrinks the intensities of marker ions
to 10% of their original intensities. This enables them to be matched when they are isobars, without the intensities
causing hit rejection when they are marker ions.
Minimum Scored Peak Intensity
Prior to performing scoring, MS/MS Search first screens the MS/MS spectrum against candidate sequences using
a simple filter. This filter is Minimum scored peak intensity. This approaches enhances search speed by
allowing candidate sequences to be rapidly and summarily rejected once a sufficient number of spectral peaks are
examined and found not to meet the threshold established by this filter.
For ultimate coverage in MS/MS Search, lower the Minimum scored peak intensity. When there are one or
more very intense peaks that overwhelm other peaks but cannot be assigned, setting this value to near 0% may improve
the number of hits at the expense of longer search times.
Guidelines: Since the matching which occurs before scoring is dependent on this filter, the value should
be set in relation to one's expectation of the quality of peak detection, i.e. noise removal and selection of
12C isotope peaks representing fragment ions corresponding to the selected Allowed Fragment-Ion types
in the spectrum prior to searching. This parameter has a very significant impact on search speed; the more unmatched
peak intensity allowed (lower percentage), the longer the search time. Composition ions are counted as unmatched
intensity, but only at 1/10 their actual peak height.
Mass Tolerances
The tolerances on both the precursor ion and fragment ions should be set to be consistent with the mass
accuracy of the instrument used to generate the data. For spectra from time-of-flight instruments, it is generally
a better idea to use units of ppm or % rather than Da, since mass accuracy is often better at lower mass than
at higher mass.
Batch Size
When you run MS/MS Search, the batch size determines the maximum number of spectra analyzed in one pass through
the database. Since all spectra of similar charge states are grouped together before splitting into batches, the
last batch for each charge state will likely contain fewer spectra than the maximum batch size.
For maximum search speed, the optimum batch size depends on the size of the database, the type of search (identity,
variable modifications, or homology), the number of modifications, and the mass accuracy of the instrument. If
the batch size is too large for the complexity of the search, the search may time out and fail to complete. For
a complex search, there is no advantage to using a larger batch size because the majority of the search time results
from the database matching rather than setting up the batches. The following table provides guidelines for the
batch size you should enter into the MS/MS Search form.
With B.04.00 and later, larger batch sizes may be specified without the risk of timeouts.
If Maximize CPUs is marked, best performance is with a batch size of 150 or more. The default batch size is
now 500. If you have less than 16 Gb of memory and are searching large data
sets, use a batch size less than 500.
If you have a Thermo Fisher Scientific Orbitrap or LTQ FT, or another instrument that produces
spectra with high mass accuracy, follow the guidelines for Agilent Q-TOF. If you have an ion trap or other
instrument that produces spectra with lower mass accuracy, follow the guidelines for Agilent ion trap.
During searches, the Spectrum Mill software dynamically reduces the batch size as the m/z increases.
For variable modifications searches and homology searches, the number of possible combinations rises dramatically
with increasing m/z; by dynamically reducing the batch size, the software reduces
memory usage.
Reversed Database Search
A reversed database search helps to rule out false positives and allows the software to calculate a
false discovery rate. If you obtain similar scores for both forward and reversed
searches, there is a higher likelihood of an incorrect assignment.
For a reversed database search, the Spectrum Mill software reverses only the internal portion of the peptide
sequences in the database rather than reversing the complete database itself. For example, the peptide:
SAMPLER
is reversed to
SELPMAR
rather than
RELPMAS.
All of these internally-reversed sequences from the database are compared to the MS/MS spectrum and the one
that returns the highest score is saved as the reversed database hit. The reversed database hit is not always
the reverse of the peptide that matched in the forward search, because a different reversed hit may score higher.
That is, all of the possible reversed hits are considered as potential matches for the experimental spectrum.
For spectra with high mass accuracy data, such as Agilent Q-TOF, many sequences will not have a reversed hit.
To Use the MS/MS Search Form
The following topics describe options available on the MS/MS Search form. In general, you should retain
the default settings, except for the options highlighted in red text on the form.
See the rest of this document for more details regarding MS/MS Search.
Search
- Start Search - Click to
place the task in the queue
for execution. The
program determines the order in which it will execute the task to do an
MS/MS search
based on the time the task entered the queue, its capacity to execute
tasks in parallel, and dependencies. Click this button after you have
either loaded the desired parameter file or manually set the
parameters. The name of the current parameter file appears in red at
the top of the form. Once you have saved a parameter file from this
form, you may start the search from a
workflow rather than manually with the Start
Search button.
- Save As - Click to save current search settings in a parameter file.
- Load - Click to load a parameter file that contains settings for MS/MS Search.
For default values, select a parameter file from the Defaults folder.
- Remove all prior MS/MS Search results - Mark this check box to remove prior MS/MS search results
for this dataset. This will also remove Spectrum Summary results.
- Maximize CPUs - Mark this check box if you want this search to take advantage of all available
CPUs (as opposed to using only a single CPU so that the other CPUs are available for other processes/users). Best performance is obtained with a Batch size of 150 or more.
If you mark this check box for a workflow, the request queue will show
two requests -- the initial one to create the batch (of files) and the
other to show the progress and search results.
Data Directories
Search Parameters
- Validation filter: Use this to search spectra having only a particular validation setting.
See Peptide Validation.
- Batch size - See Batch Size.
- Search previous hits: Mark this check box to search valid hits saved from a previous search.
See Saving Hits. If you have not yet saved the valid results using the
Tool Belt form, this search page does it for you. If you mark
the check box for Remove all prior MS/MS Search results, then the software ignores the setting
for Search previous hits (since you will have removed all previous results).
- Max. reported hits: Set to the maximum number of hits you want for each search.
- Database: Select a database. See Databases.
- Species: Choose a species if you want to narrow the search possibilities and to accelerate
searches. Please see the list of species definitions
that ship with the software, as some definitions do not encompass all possible members. Retain
the default of All to search the entire database. Be aware that because of inconsistencies
in the way species information is organized in different databases, Spectrum Mill cannot
read about 10% of the species information in NCBInr, and cannot read any of the species information in
trEMBL. See Species Filtering.
- Digest: Select the enzyme used for the proteolytic digestion. See
Enzyme Specificity / Missed Cleavages. If you select No enzyme,
then the software ignores Maximum # missed cleavages.
- Maximum # missed cleavages: Set the maximum number of missed enzymatic cleavages.
For homology searches (or searches with many variable amino acid modifications) against a full database,
reduce the number from the default of 2 to 1. See Enzyme Specificity / Missed
Cleavages.
>
Modifications
Search Criteria
The next several topics describe options available in the Search Criteria section of the MS/MS Search
form.
Matching Tolerances
- Disable match filtering (SPI, STL, S/N filter) - Mark this check box if you wish to compare
results with those from other database search engines. CAUTION: Because this mode disables signal-to-noise
and spectral quality filtering, some of the spectra you submit for the search will be poorer quality
and you will generate significantly more false positives! See Disable quality
filtering mode/disable match filtering modes. Note that the check box for Disable match filtering
is available only if it is configured in SMglobals.js. See the
server administration help for details.
- Minimum scored peak intensity: See Minimum Scored
Peak Intensity.
- Instrument: Select the instrument used for the analyses. See
Instrument / Fragment-Ion Types. For Thermo Fisher Scientific Orbitrap
and LTQ FT, see Selecting Thermo Fisher Scientific Instruments and Fragmentation Mode below.
- Masses are: See Mass Type.
- Precursor mass tolerance: See Mass Tolerances.
- Product mass tolerance: See Mass Tolerances.
- Maximum ambiguous precursor charge: Select the maximum charge state that you want MS/MS
Search to use when it encounters an extracted spectrum for which the charge state is unknown (an extracted
file that ends in *.0.pkl). While the default of +3 is optimized for CID spectra and trypsin digestion,
it may be advantageous to select higher charge states for ETD spectra or when the proteolytic enzyme
produces longer peptides (for example, the enzyme is LysC or GluC).
Spectral Quality Filtering
Certain Spectral Features calculated by the SM Data Extractor and can be used
with multiple downstream SM modules to craft a smaller subset of high value spectra.
For more details see Spectral Quality Filtering.
Search Mode
The latter three options apply only in certain homology modes.
- Calculate reversed database scores: Mark this check box if you wish to perform a database
search against peptide sequences in their forward and internally reversed directions. If you obtain similar
scores for both searches, there is a higher likelihood of an incorrect assignment. Such a search helps
to rule out false positives. See Reversed Database Search. You must
mark this check box if you want to calculate a false discovery rate (FDR), either in Autovalidation or
using the Tool Belt utility.
- Protein mobility scoring: Mark this check box if you have ion trap spectra and you wish to
have database search scores take into account the likelihood of fragmentation at a particular site, based
upon relative proton mobility and other factors. See Ranking/Scoring of Results.
Do not mark this check box if you have peptides that are modified by iTRAQ, lysine mass tagging, guanidination,
or phosphorylation, because these modifications change the expected fragmentation pattern.
- Dynamic peak thresholding: Mark this check box if you wish to use a scoring enhancement that
enables identification of more low-abundance and short-chain peptides. For each extracted spectrum, the
software calculates the search scores as the number of spectral peaks varies from n=4 up to the maximum
set by the variable peakLimitCount in instrument.txt. It then displays the best score
from the set.
- Discriminant scoring: The
default selection is "Off", which disables the creation of the files that are
necessary for use of the Autothresholds – discriminant strategy
in autovalidation. The second selection is "Score only." It
replaces the "Disable" field from the prior release and is here for
backwards compatibility. However, "Score only" negates
the purpose of discriminant scoring, which is to use a combination of
other factors in the score. If you choose one of the selections
below, discriminant scoring is enabled and uses the calculated set of
coefficients for the Agilent instrument selected, or for an instrument
that you choose. You can create a set of coefficients for an
instrument of your choosing with the Tool Belt utility. After you
do this, the name of the set of coefficients appears in the selection
list.
- Agilent QTOF standard - Discriminant scoring will use this set of already calculated coefficients for the Agilent QTOF.
- Agilent
Ion Trap CID standard - Discriminant scoring will use this set of
already calculated coefficients for the Agilent ion trap CID
fragmentation mode.
- Agilent Ion Trap ETD standard -
Discriminant scoring will use this set of already calculated coefficients for the Agilent ion trap ETD fragmentation mode.
- Search mode: Select a search mode. See Search Mode. If you
select the variable modifications or one of the homology modes, the software displays one or more of
the following options:
- Precursor mass shift or Precursor mass shift range: The default setting depends
on which homology mode you use. See Precursor Mass Shift. In the
Variable modifications search mode, set a number that is appropriate for the types and numbers of
modification(s) that you are searching. Larger values may produce longer searches and increase the possibility
of false positives.
- Single AA substitution / Unassigned single mass gap - In one of the homology modes, if you
wish to search for a single amino acid substitution, select Single AA substitution. If you wish
to instead search for an unknown modification, select Unassigned single mass gap.
Data Files
- Fragmentation Mode - Select one of the modes below for
an ion trap Instrument.
These selections take the place of the ion trap "MIX" Instrument
selections found in versions previous to B.04.00. For Agilent
Q-TOF and other instruments that only acquire CID, always specify All.
- CID - collision-induced dissociation via resonance excitation in the ion trap
- ETD - electron transfer dissociation in the ion trap
- ETHCD - electron transfer dissociation in the ion trap, transfer all ions to the HCD collision cell, dissociate via beam-type collision induced dissociation.
- HCD - isolate precursor ions in the ion trap or with Q1, dissociate in the HCD collision cell via beam-type collision induced dissociation, scan out the fragment ions and detect in the Orbitrap
- PQD - pulsed-q dissociation - isolate precursor ions and dissociate in the ion trap, but without the low-mass cutoff that you see with CID spectra
- Spectrum Files - Modify this list if you want to process only a subset of the files in the
data directory. Wildcards (*) are supported.